Cell culture
U-2 OS cells (ATCC) were grown in DMEM (Corning 10–013-CV) supplemented with 10% FBS (ThermoFisher, 26,140,079), 1X GlutaMAX (ThermoFisher, 35,050), 1X antibiotic–antimycotic (ThermoFisher 15,240) and 1X Non-Essential Amino Acids (ThermoFisher, 11,140,050). Cells were plated at 30% confluence (Day -2) and grown to reach confluence (2 days later, day 0) or allowed to continue to grow to become over-confluent (2 additional days, day + 2).
Cytoplasmic extraction, nuclear extraction and chromatin purification
Isolation of cytoplasmic extracts, nuclear extracts, and purified chromatin was performed as previously described [47]. Cells were harvested, washed in PBS, and resuspended in low salt buffer (LSB) (20 mM HEPES pH 7.9, 25% glycerol, 1.5 mM MgCl2, 2 mM EDTA, 1 mM DTT, Halt Protease and Phosphatase Inhibitor Cocktail). Cells were incubated on ice for 15 min to allow the cells to swell. Cells were lysed by adding the non-ionic detergent NP-40 (0.75% final) and gently passed through a 21-gauge needle ten times. Nuclei were collected by spinning at 1100 × g at 4 °C for 5 min. The supernatant was collected as the cytoplasmic extract. Nuclei were washed twice with LSB and then resuspended in 500 µLs of LSB. The pelleted nuclear volume (PNV) was determined by measuring the total volume—500 µL LSB. Nuclei were repelleted and resuspended in ½ PNV of the LSB. ½ PNV of high salt buffer (HSB) (20 mM HEPES pH 7.9, 25% glycerol, 1.5 mM MgCl2, 1.6 M NaCl, 1 mM DTT, Halt Protease and Phosphatase Inhibitor Cocktail) was added dropwise while vortexing at a low speed to achieve a final NaCl concentration of 400 mM. Samples were incubated at 4 °C while shaking for 1 h before centrifuging them at 21,000 × g for 10 min at 4 °C. The supernatant was collected as the soluble nuclear extract. The pellet (chromatin fraction) was washed twice for 10 min with shaking the 400 mM NaCl HSB, pelleted, and resuspended in 1X SDS-PAGE load dye (final: 50 mM Tris–HCl pH 6.8, 3% SDS, 10% glycerol, 5% β-mercaptoethanol, 0.002% bromophenol blue). Samples were boiled at 95 °C for 5 min and cooled on ice three times before shearing the chromatin by sonication (15 s using the Misonix Sonicator3000 (Power setting 3.0), repeated until pellet dissipated completely). The cytoplasmic and nuclear soluble extracts were quantitated using the BCA Protein Assay Kit (ThermoFisher 23,225) prior to loading for Western analysis.
Western blot analysis
Chromatin samples were fractionated on a 15% SDS-PAGE gel (cast in house), transferred to a 0.2 µm nitrocellulose membrane (GE Healthcare, GE10600001) using Towbin buffer (25 mM Tris pH 8.8, 192 mM Glycine, 20% methanol) on a Hoefer TE 77 semi-dry transfer unit for 1.5 h at 55 mA/membrane. 20–30 µgs of cytoplasmic and nuclear soluble extracts were fractionated on a 10% SDS-PAGE gel and transferred to a 0.45 µm nitrocellulose membrane using the same buffer and transfer unit. Each membrane was incubated in a blocking solution (4% non-fat milk in TBS) for 1 h prior to replacement with primary antibody diluted in 1.5% milk in TBS. Antibodies and the concentrations used are supplied in Additional file 9: Table S6. Membranes were incubated while rocking in the primary antibody overnight at 4 °C, washed in TBS-T (TBS, 0.1% Tween-20) three times for 10 min and incubated in TBS-T with 1.5% milk with a secondary antibody for 45 min while covered at room temperature. The membrane was then washed three times for 10 min in TBS-T, rinsed in diH2O and imaged at 700 nm using the Odyssey® CLx imaging system (LI-COR). Equivalent loading was confirmed by staining the membrane following imaging with Amido Black stain (10% acetic acid, 0.1% Amido Black 10B (w/v)).
Viral plasmid transfections
Half a million 293 T cells (ATCC) were plated in a 6-well plate 24 h prior to transfection. A transfer plasmid, psPAX2, and pMD2.G were transfected at a 4:3:1 ratio into the 293 T cells using Lipofectamine 3000 according to the manufacturer’s protocol (ThermoFisher, L3,000,001). Fresh media was replaced the following day, and after 72 h media containing virus was collected.
Lentivirus transductions
200 k U2OS cells were plated in a 6-well plate 24 h prior to transduction. Virus containing media was filtered using a 0.45 µm cellulose acetate membrane (VWR 28145–481) and 10 µg/mL of Polybrene (Millipore, TR-1003-G) was added. Viral media was added to the cells and a spinfection performed at 1100 × g for 1 h at 33 °C. Fresh media was added 24 h later. Puromycin was added at a concentration of 2 µg/mL for 5 days with replacement every 2 days. shRNA plasmids were obtained as glycerol stocks from Sigma and grown for midipreps using the PureLink HiPure Plasmid Midiprep Kit (ThermoFisher, K210004). The following shRNA’s were used in this study: MMP2 (TRCN0000051523 and TRCN0000051526), CTSB (TRCN0000003655 and TRCN0000003658), and pLKO.1 control (SHC001).
RNA isolation
Total RNA was extracted using TRIzol reagent according to the manufacturer’s protocol (ThermoFisher, 15,596) Briefly, cells were washed with PBS briefly before adding 1 mL of TRIzol directly to cells. Cells were scraped and the mixture transferred to a microfuge tube. Chloroform was added at a ratio of 1:5 (v/v) and the samples were vortexed vigorously and centrifuged at maximum speed at room temperature for 15 min. The RNA-containing aqueous phase was isolated and combined 1:1 (v/v) with Isopropanol at room temperature. RNA was pelleted at max speed at 4 °C and washed twice with 70% ethanol. The dried pellet was then resuspended in nuclease free diH2O and RNA concentration determined by spectrophotometry.
RT-qPCR
One µg of total RNA was reverse transcribed using the SuperScript IV VILO Master Mix according to the manufacturer’s protocol (ThermoFisher, 11,756). 10 ng’s of the resulting cDNA was used for qPCR using the PerfeCTa® SYBR® Green FastMix® (QuantaBio, 95,072)) according to the manufacturer’s protocol in an iQ5 iCycler (BioRad). 5 µM each of the forward and reverse primer were used (see Additional file 9: Table S7). Quality and specificity of primer sets were validated by melt curve analysis. Expression of each gene was calculated as 2^(− ∆∆Ct) relative to the GAPDH housekeeping gene. Three independent biological replicates were performed to determine the average expression change and standard deviation.
In vitro H3 cleavage assay
Nuclear soluble extracts (3 μgs) or recombinant protein (100 ngs) were incubated with acid extracted core histones purified from C2C12 myoblasts (1 μg) in Cleavage Buffer (10 mM Tris–HCl pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM CaCl2) at 37 °C for 1 h [23]. Reactions were quenched with 6X SDS-load dye and boiled for 5 min. SDS-PAGE and Western analysis was performed as described above.
RNA-seq
RNA-seq was performed by Novogene Corporation. Total RNA quality was assessed using an Agilent TapeStation RNA ScreenTape (Agilent, 5067–5576) and only samples with a RIN score above 7 were used for sequencing. mRNA was selected using the NEBNext Poly(A) mRNA Magnetic Isolation Module (NEB, E7490), and cDNA synthesis and libraries made using the NEBNext Ultra II RNA Library Prep Kit for Illumina (NEB, E7770) according to the manufacturers protocol. Libraries were sequenced on a NovaSeq 6000 System using an S1 Flow Cell (Novagene Corporation) in PE 2 X 150 bp mode. Over 20 million reads were obtained for each sample.
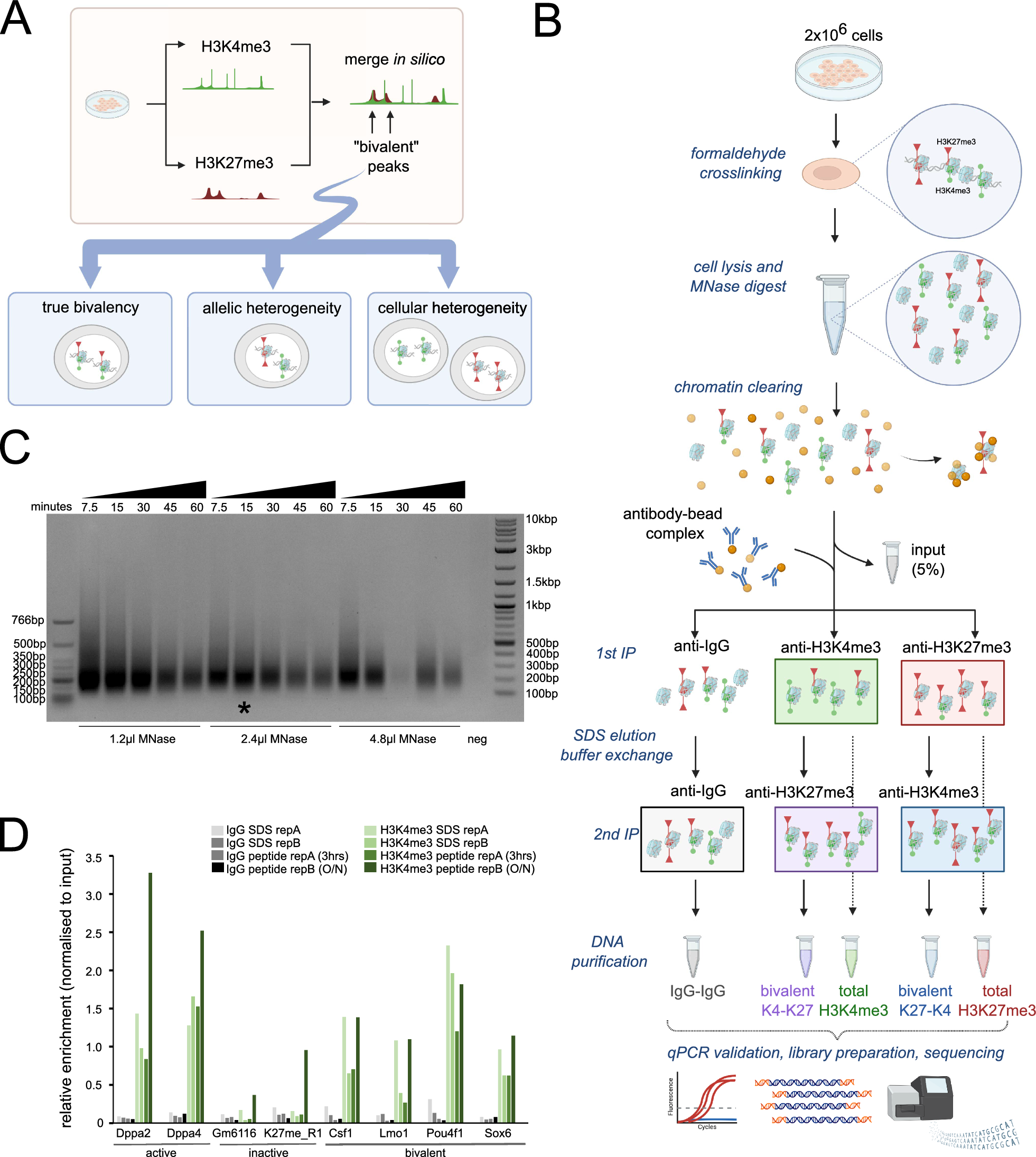
MNase digestion of chromatin
Native ChIP-seq was adapted from [48]. 2.85 Units of micrococcal nuclease (MNase) per million nuclei from Worthington (LS004797) was used for all experiments. Briefly, cells were washed with ice-cold PBS before scraping and collecting them. Cells were kept on ice and all spins done at 4 °C unless otherwise noted. All buffers had Halt Protease and Phosphatase Inhibitor Cocktail and 10 mM Sodium Butyrate added. Cells were resuspended in native ChIP Lysis Buffer (NLB) (10 mM Tris–HCl pH 8.0, 10 mM NaCl, 3 mM MgCl2, 0.1% NP-40) and lysed by gently passing them through a 21-gauge needle ten times. Nuclei were collected by spinning at 1100 × g for 5 min, and supernatant discarded. Nuclei were resuspended in NLB, quality assessed and counted using 0.2% Trypan Blue (ThermoFisher, 15,250,061). 40 million nuclei per cell line were resuspended in 500 µLs of MNase Digestion Buffer (MDB) (10 mM Tris–HCl pH 8.0, 10 mM NaCl, 3 mM MgCl2, 0.1% NP-40, 250 mM Sucrose, 3 mM CaCl2). Nuclei were incubated at 37 °C for 5 min, and then MNase added (2.85 U/1 million nuclei). Nuclei were incubated at 37 °C and lightly vortexed every minute for 4 min. MNase was quenched using EDTA (4 mM final concentration) and nuclei incubated on ice for 10 min. Nuclei were pelleted at 1100 × g for 5 min and the supernatant saved as the soluble 1 (S1) fraction. The remaining nuclei were resuspended in 300 µLs of native ChIP release Buffer (NRB) (10 mM Tris–HCl pH 7.5, 10 mM NaCl, 0.2 mM EDTA) and incubated on a nutator for 1 h at 4 °C. The nuclei were centrifuged at max speed for 10 min at 4 °C and the supernatant collected as the soluble 2 (S2) fraction. The remaining pellet (P) was washed once with NRB and then frozen for further analysis.
Native ChIP
The S1 and S2 fractions were combined and diluted 10X in native ChIP Incubation Buffer (NIB) (10 mM Tris–HCl pH 7.5, 70 mM NaCl, 2 mM MgCl2, 2 mM EDTA, 0.1% Triton X-100). The lysate was pre-cleared with 60 µL of Pierce ChIP-Grade Protein A/G Magnetic Beads (ThermoFisher, 26,162) with rotating at 4 °C for one hour. Beads were discarded and lysate divided up. Histone ChIPs used 50 µgs of chromatin (determined from collecting S1/S2 pooled input and isolating DNA for quantification) and HA ChIP used 200 µgs of chromatin. Antibodies were added for each IP (See Additional file 9: Table S6 for amounts used) and rotated end-over-end overnight at 4 °C. 25 µLs of magnetic beads per IP were washed 3X in NIB and blocked in 1% BSA overnight. The following morning beads were washed 3X with NIB and resuspended in their original volume. 25 µLs of blocked beads were added to each IP for three hours. Each lysate was washed 4X in native ChIP Wash Buffer (NWB) (20 mM Tris–HCl pH 7.5, 135 mM NaCl, 2 mM EDTA, 0.1% Triton X-100) with 5 min of rotation at 4 °C between each wash. Lysates were then washed 2X in native ChIP LiCl Wash Buffer (LWB) (10 mM Tris–HCl pH 7.5, 250 mM LiCl, 1 mM EDTA, 0.5% NP-40, 0.5% Na-DOC) with 5 min of rotation at 4 °C. Lysates were then washed once in TE Wash Buffer (TWB) (50 mM Tris–HCl pH 8.0, 10 mM EDTA), followed by a short spin to get rid of all residual buffer. Beads were resuspended in 200 µL of Elution Buffer (EB) (1% SDS, 0.1 M NaHCO3) and shaken at 800 RPM at 37 °C for 30 min. A final elution of 10 min at 65 °C was then performed. Supernatant was collected and RNase A (2 µgs, ThermoFisher, R1253) and Proteinase K (5 µgs, VWR, E195) were added and incubated at 37 °C for 30 min and then 55 °C for 3 h. 200 µL of Phenyl:Chloroform:Isoamyl Alcohol (25:24:1, Sigma, P2069) was added and samples vortexed for 15 s, and then centrifuged at max speed at room temp for 15 min. The aqueous phase was collected and 2.5X volumes 100% Ethanol, 10% (v/v) 3 M Sodium Acetate, and 3 µgs of Glycogen (VWR, N632) were added, vortexed, and incubated at − 80 °C for 1 h. Samples were centrifuged for 30 min at max speed at 4 °C, washed with 70% ethanol, and re-pelleted. Dried pellets were resuspended in 50 µL of nuclease free water. DNA was stored at − 20 °C until used for library prep or qPCR.
ChIP library prep
Isolated ChIP DNA was used to make libraries using the NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB, E7645S). Libraries were constructed according to the manufacturers protocol. Briefly, DNA was subjected to end-repair and dA-tailing. NEBNext adaptors (0.6 µM) were ligated onto DNA fragments, and then U excision performed by the USER enzyme. Size selection was performed using AMPure XP (Beckman Coulter, A63880) to get rid of additional adaptors. Amplification was performed using the NEBNext Ultra II Q5 Master Mix, the universal i5 primer and one index i7 primer. The appropriate cycles were determined by including 1X EvaGreen Dye (Biotium, 31,000) and stopping the reaction once the library reached 3000 RFU. Following PCR, a double-sided side selection was performed using AMPure XP beads (0.9–0.55) to get rid of residual primers and any large DNA fragments. Library quality and concentration was assessed using an Agilent Bioanalyzer and the HS DNA Kit (Agilent, 5067–4626). Libraries were sequenced on a NovaSeq 6000 System using an S1 Flow Cell (Novagene Corporation) in PE 2 X 150 bp mode. Over 30 million reads were obtained for each library.
ChIP qPCR
1 µL of isolated ChIP DNA was used with 500 nM primers (both forward and reverse) and with PerfeCTa® SYBR® Green FastMix® (QuantaBio, 95,072) according to the manufacturer’s protocol in an iQ5 iCycler (BioRad). All primer sequences can be found in Additional file 9: Table S8. Fold change over H3 was calculated as 2^-(IPct-H3IPct). 4 technical replicates were performed for each biological replicate. At least two biological replicates were done for each ChIP-qPCR. Statistical significance was calculated using a student T-test in Prism Graph Pad.
Sequencing data analysis
All ChIP-seq and ATAC-seq data was processed through a modified version of the ENCODE pipeline (https://www.encodeproject.org/data-standards/chip-seq/) [49]. MACS2, bedtools and deepTools were used for peak calling, comparing datasets and visualization respectively [50,51,52]. Detailed methods of all analysis is provided in the supplemental methods. RNA-seq data was analyzed using DEseq2 and RSEM according to their documentation [53, 54]. ggplot2 was used for making volcano, violin, and boxplots [55]. Tidyverse and dyplr were utilized for merging ChIP-seq and RNA-seq datasets [56, 57]. Detailed methods of all analysis are provided in the supplemental methods Additional file 10.




留言 (0)