
記住我

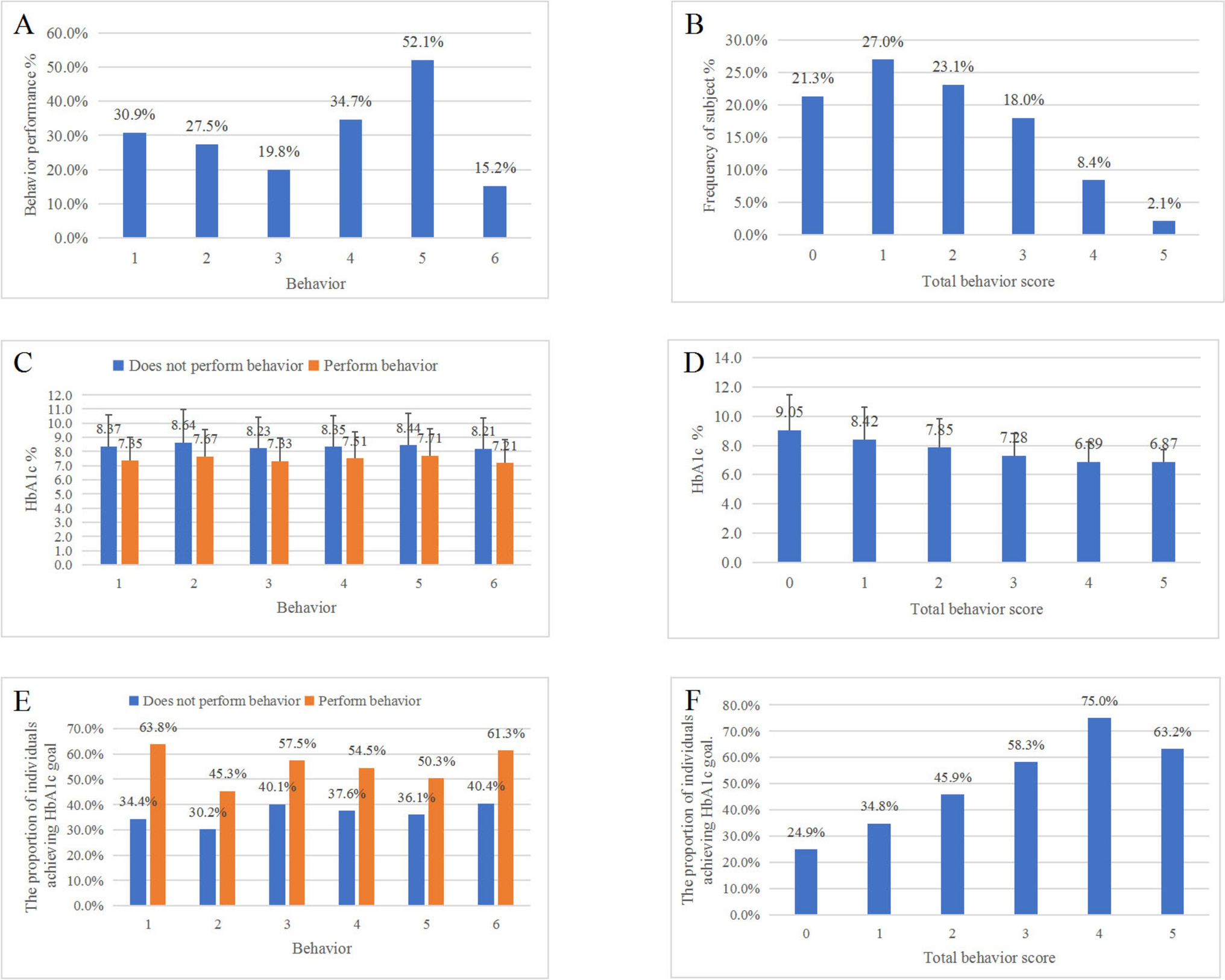

To elucidate the proteomic profile of PTMC, six paired normal tumor tissues were collected and further sequenced using TMT-labeled LC–MS /MS. Depending on the status of lymphatic metastases, six paired samples were divided into N0 and N1 groups. The N1 group included patients with lymphatic metastases, while the N0 group included patients without lymphatic metastases. In this study, we paid more attention to the following two differences: one was the difference between tumor and normal (tumor/normal group), and the other was the discrepancy between the N1 group and the N0 group (N1/N0 comparison group).
In this study, a total of 5203 proteins were identified and quantified with a fold change threshold of > 1.30 or < 0.67. In the tumor/normal group, 487 proteins were upregulated and 486 proteins were significantly downregulated compared with the normal tissues. Similarly, more than 200 proteins were upregulated and downregulated in the N0 tumor/N0 normal group and the N1 tumor/N1 normal group, respectively. However, contrary to our expectations, only 20 DEPs were observed in the N1 tumor/N0 tumor group, and 53 DEPs were detected in the standardized N1 tumor vs normal/N0 tumor vs normal group (Fig. 1A). To further investigate the possible DEPs between tissues with lymph node metastasis and negative tissues, we considered the following three comparisons: the standardized N1 tumor vs. normal/N0 tumor vs. normal group, the N1 tumor/N0 tumor group, and DEPs between N1 tumor/N1 normal and N0 tumor/N0 normal. Principal component analysis showed a clear distinction between tumor and normal samples, but an overlap between N1 tumor and N0 tumor (Fig. 1B). As shown in Fig. 1C, hierarchical cluster analysis revealed a clear differential expression pattern between tumor and normal samples. However, when comparing N0 tumor and N1 tumor, the difference was not as clear as when comparing tumor and normal, but a slight change was also observed.
Fig. 1
Schematic representation of the overall study design and differentially expressed proteins between Tumor and Normal tissues using liquid chromatography–tandem mass spectrometry (LC–MS/MS) analysis. A The bar chart shows the number of differentially expressed proteins (DEPs) in each functional group. B Principal component analysis (PCA) plot showing unsupervised clustering among the 12 samples that combined Tumor and Normal tissues. C Heat map representation of total expressed proteins in all comparison groups. Significantly enriched downregulated (green) and upregulated (red) proteins. The dendrograms represent the classification of proteins
The dynamic expression patterns and associated pathways in the normal-tumor-metastasis progression of PTMCTo analyze the dynamic expression patterns of DEPs during the progression of PTMC, the 5203 DEPs were divided into 6 patterns (clusters 1 to 6) using Mfuzz analysis. As shown in Fig. 2A, DEP clusters were enriched according to progression status, followed by normal tissue (N0 normal and N1 normal), tumors without lymphatic metastases (N0 tumor), and tumors with lymphatic metastases (N1 tumor). The expression patterns of clusters 1 and 3 gradually increased with disease progression. These DEPs were significantly enriched in Parkinson’s disease, Huntington’s disease, myocardial contraction, apoptosis, and Wnt signaling. Meanwhile, clusters 4 and 5 were initially downregulated during the transition from normal to tumor, but were upregulated again during the transition from N0 tumor to N1 tumor. These related DEPs were mainly enriched in Hedgehog signaling, fluid shear stress, atherosclerosis, African trypanosomiasis, and complement and coagulation. This suggests that these DEPs, as well as their associated signaling pathways, may play an important role in the progression of PTMC.
Fig. 2
Results of the clustering analysis using Mfuzz and pathways enrichment analyses highlighting the significance of differentially expressed proteins (DEPs) using the Kyoto Encyclopedia of Genes and Genomes (KEGG) and protein–protein interaction (PPI). A Mfuzz clustering analysis of DEPs. Six clusters are identified using Mfuzz software. Proteins in clusters 1 and 3 show a consistently increasing trend from Normal tissues to N0-Tumor tissues which represent the appearance of papillary thyroid microcarcinoma (PTMC). Proteins in clusters 4 and 5 show a consistently decreasing trend from N0-Tumor to N1-Tumor tissues, which represent the progression of PTMC. B The KEGG pathway enrichment analysis of DEPs. The left pie chart shows the top 10 distribution of pathways in each KEGG category of the Tumor/Normal comparison group. The right panel shows the top 10 KEGG pathways in the N1/N0 comparison group. C PPI network construction. The left PPI network is based on the proteins enriched in apoptosis, thyroid hormone synthesis, cholesterol metabolism, lysosome, pyruvate metabolism, and ribosome, which are selected from the top 10 pathways in the Tumor/Normal KEGG pathway and important pathways in previous research. The right PPI network is based on the proteins enriched in ribosome, pyruvate metabolism, cholesterol metabolism, tricarboxylic acid (TCA) cycle, and PI3K-Akt signaling pathway, which are selected from the top 10 pathways in the N0/N1 comparison group and important pathways in previous research. The proteins marked with a bigger size are selected for parallel reaction monitoring (PRM) and immunohistochemical (IHC) protein analyses
To investigate the specific functions of DEPs, KEGG pathway analysis was performed. As shown by the circle map in Fig. 2B, DEPs in the tumor/normal group were mainly enriched in ribosome, lysosome, phagosome, cholesterol metabolism, and thyroid hormone synthesis. Similarly, DEPs from the N1 vs. N0 comparison group were mainly enriched in AGE-RAGE signaling pathway, cholesterol metabolism, pyruvic acid pathway, etc. In addition, PPI was used to investigate the possible network interactions of the DEPs (Fig. 2C). The DEPs from the tumor/normal group interacted and were enriched in the following metabolic pathways: apoptosis, lysosome, ribosome, cholesterol metabolism, pyruvate metabolism, and thyroid hormone synthesis. Similarly, DEPs from the N1/N0 comparison group interacted and were enriched in the tricarboxylic acid (TCA) cycle and PI3K-Akt pathway. Based on the above analysis and literature review, 20 candidate proteins were selected as targets for validation.
Validation of targeted proteinsTo validate the targeted proteins, 20 paired samples were collected from PTMC and further analyzed with PRM. The basic clinical information of the patients is shown in suppletment Table 1. Similarly to the original PTMC sequencing, half of the patients had lymph node metastases (N1 stage) in the validation, but only two out of ten were in the progressive stage (N1b stage). Indeed, the heat map of PRM showed an obvious trend and pattern that changed according to the progression normal-tumor-metastasis (Fig. 3A). The detailed statistical results showed that 18 of the 20 target proteins were consistent with the previous sequencing data (Fig. 3B). Unfortunately, the differential expression of DEPs between N1- and N0-stage patients was not obvious, which may be due to the lower proportion of N1b-stage patients. In addition, immunohistochemistry (IHC) was performed to investigate the different expression levels. As shown in Fig. 3C, most results were consistent with PRM. Take NPC2 and PHDB as examples, those two genes were higher expressed in PTC tissues, compared with the thyroid gland. The association between target proteins and clinical features needs to be further validated using a larger number of samples.
Fig. 3
Confirmation of proteomic alterations using parallel reaction monitoring (PRM) and immunohistochemical (IHC) protein analyses. A Heatmap representation of the overall abundance profile of all 20 detected proteins in the 20 PRM pairs of papillary thyroid microcarcinoma (PTMC) tissues and adjacent noncancerous tissues. Red represents upregulation, and blue represents downregulation. B Violin plots represent the significantly different expression levels of PRM proteins between the Tumor/Normal comparison group and the N0/N1 comparison group. One asterisk indicates P < 0.05–0.01; two asterisks indicate P < 0.01–0.001. C IHC expression of PRM protein between PTC tissues and gland tissues
Predictive performance of biomarkers calculated via machine learningFurthermore, using machine learning strategies based on the PRM results, we evaluated the predictive power of the validated proteins (Fig. 4). In distinguishing between tumor and normal nodules, the following five proteins showed the best performance: P50479 (PDLIM4), P04083 (ANXA1), P14618 (PKM), P61916 (NPC2), and P02545 (LMNA). All AUCs of these five proteins were greater than 0.9 (Fig. 4A and B). In addition, we further established a possible clinical predictive model by machine learning based on the PRM sequencing result. As illustrated in Fig. 4C, P50479 (PDLIM4) together with P04083 (ANXA1) could distinguish benign nodule from malignant nodule, and the AUC was as high as 1.00. Similarly, we evaluated the predictive power of the validated proteins to distinguish patients with lymph node metastases (N1) from patients without lymph node metastases (N0). Among them, P02751 (FN1) was the most relevant protein with an AUC of 0.690 (Fig. 4D and E). These results indicate its potential role as a biomarker.
Fig. 4
Machine learning model to identify potential target proteins of onset and progression in papillary thyroid microcarcinoma (PTMC). A Overall distribution of feature scores among proteins in normal and tumor tissues in the training cohort. B The best performance of the top five proteins for predicting normal and tumor tissues of PTMC. C Machine learning results predicting tumor from normal tissues of PTMC. D Overall distribution of feature scores among proteins in N0/N1 comparison group in the training cohort. E The performance of FN1 for predicting metastasis of PTMC
Performance of the five-protein panel to predict prognosisTo investigate their prognostic value in thyroid cancer, we further evaluated 18 validated proteins from the TCGA database. Interestingly, five proteins showed a potential prognostic role: FN1, IDH2, VDAC1, FABP4, and TG (Fig. 5). First, FN1, IDH2, and VDAC1 were more highly expressed in thyroid cancer tissues than in normal tissues; however, FABP4 and TG were less highly expressed in thyroid cancer tissues (Fig. 5A). Consequently, the higher expression of FN1, IDH2, or VDAC1 meant a worse prognosis, as indicated by the 5-year progression-free interval (PFI). In contrast, lower expression of FABP4 and TG showed a worse prognosis (Fig. 5B). Moreover, the expression of the five proteins was associated with some clinicopathologic features, such as simplified tumor stage, extra-thyroidal carcinoma, and histologic type (Fig. 5C). To comprehensively assess the prognosis of a thyroid cancer patient, we attempted to construct a nomogram depending on the expression of these five proteins based on the above analysis (Fig. 5D). For example, a thyroid cancer patient with high FN1 risk (51 points) and high VDAC1 risk (100 points) received a total score of 151, and the 1-, 3-, and 5-year survival rates were 96%, 90%, and 85%, respectively. As shown in Fig. 5E, the predictive accuracy of this nomogram was good, as indicated by the higher C-index of 0.685 (confidence interval: 0.645–0.726). These results indicate that the five-protein nomogram has good predictive and prognostic performance. The schematic workflow of the study was shown in Fig. 6.
Fig. 5
The potential target proteins showed different clinical characteristics in The Cancer Genome Atlas (TCGA) and established a nomogram for predicting the probability of 5-year progression-free interval (PFI) for patients with papillary thyroid carcinoma (PTC). A Expression levels of FABP4, FN1, IDH2, TG, and VDAC1 in TCGA thyroid cancer cohort with normal and tumor tissues were analyzed. B Survival curves of PFI between FABP4, FN1, IDH2, TG, and VDAC1-high and -low patients with PTC. C Relationships between FABP4, FN1, IDH2, TG, and VDAC1 and clinicopathological features. D A nomogram for predicting the probability of 5-year PFI for patients with PTC. E Calibration plots of the nomogram for predicting the probability of PFI at 5 years and the relationship between risk score and clinical information
Fig. 6
The schematic workflow of the study
留言 (0)