
記住我










Alzheimer’s disease (AD) is a neurodegenerative disease that causes a progressive decline in memory and cognitive function.1 Although the majorities of AD cases occur on a sporadic basis, mutations in three genes, including amyloid precursor protein (APP), presenilin 1 (PSEN1) and presenilin 2 (PSEN2), can lead to rare familial AD. Its symptoms occur earlier than sporadic AD, usually between 30 years and 50 years of age. However, late-onset AD, more than 65 years of age onset, may be motivated by a complex interaction between genetic and environmental factors.2 The current theories of AD pathogenesis are β-amyloid (Aβ) plaques and neurofibrillary tangles, with consequences of microglial activation, synaptic deficiency and neuronal loss.1 3
The GSN gene, encoding gelsolin (GSN), is a calcium-regulated actin regulatory protein that is recognised to be functionally involved in inflammation, cell movement, apoptosis and cancer development.4 The GSN is suggested to be implicated in AD, based on the previous findings that GSN can bind Aβ, inhibit Aβ-induced toxicity, and protect cells from apoptosis and reactive oxygen species (ROS).4–6 The well-known pathogenic missense mutations of the GSN gene have been reported to be associated with familial amyloidosis of the Finnish type (FAF), which mainly manifests as corneal lattice dystrophy, cranial neuropathy, peripheral neuropathy and cutis laxa.7 Recently, we reported patients with GSN frameshift mutations were characterised by the typical AD phenotype.8 Among the five patients with AD (cases 1–5) with GSN frameshift mutations, unfortunately, only the patient with the K346fs mutation (case 1) accepted Pittsburgh compound ([11C]PIB)positron emission tomography (PET) examination and was confirmed to have cerebral Aβ deposition; however, no patient agreed to undergo skin biopsy, which is of great help in differentiating the diagnosis of FAF.8 Thus, we could not completely determine whether these patients with frameshift mutations had FAF. Therefore, the heterogeneity of genotype–phenotype in the GSN gene, as well as the role of the GSN frameshift mutation in the pathogenesis of AD, is still unclear.
In the present study, we found 0.50% of AD cases carrying the GSN frameshift mutations, who were not accompanied by other dementia-related gene mutations through whole genome sequencing (WGS) technology. The diagnosis of AD, but not FAF, was confirmed by [11C] PiB-PET and skin biopsy. Besides, the GSN loss-of-function effect of frameshift mutations was verified through in vitro experiments (Aβ-induced toxicity and apoptotic tests) and the detection of decreased GSN mRNA and GSN level in plasma. Moreover, the GSN level in plasma was increasing in sporadic AD cases compared with controls, indicating that the increasing plasma GSN may be a potential biomarker for AD. This work first claimed that the frameshift mutations in the GSN gene were associated with sporadic AD through its loss-of-function mechanisms.
Materials and methodsSubjectsWe enrolled a total of 1192 patients with AD from the Department of Neurology, Xiangya Hospital (table 1). All patients were diagnosed as AD according to the 2011 National Institute of Ageing and Alzheimer’s Association (NIA-AA) criteria for probable AD.9 All participants were sequenced through WGS to identify the GSN mutational spectrum, and meanwhile, other dementia-related gene causative mutations were excluded. The GSN mutations were further validated in two validation cohorts of the Cognitive Impairment Multicenter Database and Collaborative Network in China (CI-MDCNC), as well as the Alzheimer’s Disease Sequencing Project (ADSP)10–12 (online supplemental methods). The CI-MDCNC includes 884 patients with probable AD who underwent gene-targeted sequencing (online supplemental methods). Besides, 1403 cognitively unimpaired controls were enrolled from Xiangya Hospital Health Management Centre (table 1).
Table 1Demographic information of the enrolled gene sequencing and plasma GSN detection participants
Additionally, 124 patients with AD and 103 cognitively unimpaired controls, which were sex-matched and age-matched, were recruited for GSN detection in plasma samples. A total of 82 patients with AD and 59 controls further underwent cerebrospinal fluid (CSF) GSN detection. Meanwhile, 82 patients with AD underwent CSF AD biomarker examinations, including Aβ40, Aβ42, Aβ42/40, t-tau and p-tau, which met the 2018 NIA-AA criteria13 for the framework diagnosis of AD for A+T+n+classification (online supplemental methods).
WGS and data analysisThe formalin-fixed paraffin-embedded (FFPE) DNA was extracted using the MagPure FFPE DNA LQ Kit following the manufacturer’s instruction. The sample’s concentration was detected by Qubit fluorometer. The integrity and purification of samples were detected by agarose gel electrophoresis. One nanogram and more FFPE DNA were randomly fragmented by Covaris. Fragmented DNAs were tested by Agilent 2100 and purified by the Agencourt AMPure XP kit. The selected fragments were end-repair and 3′ adenylated; at the same time, the adapters were ligated to the ends of these 3′ adenylated fragments. These fragments were amplified with KAPA HiFi HotStart DNA polymerase, and the PCR products were purified with the Agencourt AMPure XP kit. The library was qualified by the Agilent 2100 Bioanalyzer and ABI StepOnePlus real-time PCR (RT-PCR) system. The qualified libraries were sequenced pair end on the Hiseq4000/Xten/Novaseq system (BGI, Shenzhen, China). We used Genome Analysis Toolkit software to detect (Single Nucleotide Variants) SNVs and indels. All SNVs and indels were filtered and estimated via multiple databases, including Genome Aggregation Database and Exome Aggregation Consortium. Common variants and rare variants were classified according to minor allele frequencies (MAFs; common variants: MAF ≥0.01, rare variants: MAF <0.01).14
Real-time PCRThe blood samples of subjects were freshly collected. Total RNA was extracted using TransZol Up Plus RNA Kit (catalogue number 315–150; GeneAll, Korea), and cDNA was synthesised from 1 µg of total RNA by using the NovoSceipt Plus All-in-One First Strand cDNA Synthesis Kit (catalogue number E047; Novoprotein, China). For PCR, the cDNA was amplified with 2× M PCR OPTI Mix (catalogue number B45012; Bimake, USA) in a T100 Thermal Cycler (Bio-Rad, USA). The primers for the two GSN frameshift mutations are shown in online supplemental figure S1A. Next, the PCR products were subjected to agarose gel electrophoresis. For RT-PCR, the cDNA was amplified with 2× SYBR Green qPCR Master Mix (catalogue number B21202, Bimake) using FTC-3000 RT-PCR system (Funglyn Biotech, Canada). The relative standard curve method (2–ΔΔCt) was used to determine the relative gene expression, and GAPDH was used as a housekeeping gene for internal normalisation. The PCR primers were as follows: GSN: forward, 5′-AGCTGGCCAAGCTCTACAAG-3′, and reverse, 5′-TTCCTCTCCTCCGTGTTTGC-3′; GAPDH: forward, 5′-AGTTAAAAGCAGCCCTGGTGA-3′, and reverse, 5′-TCGACAGTCAGCCGCATCT-3′.
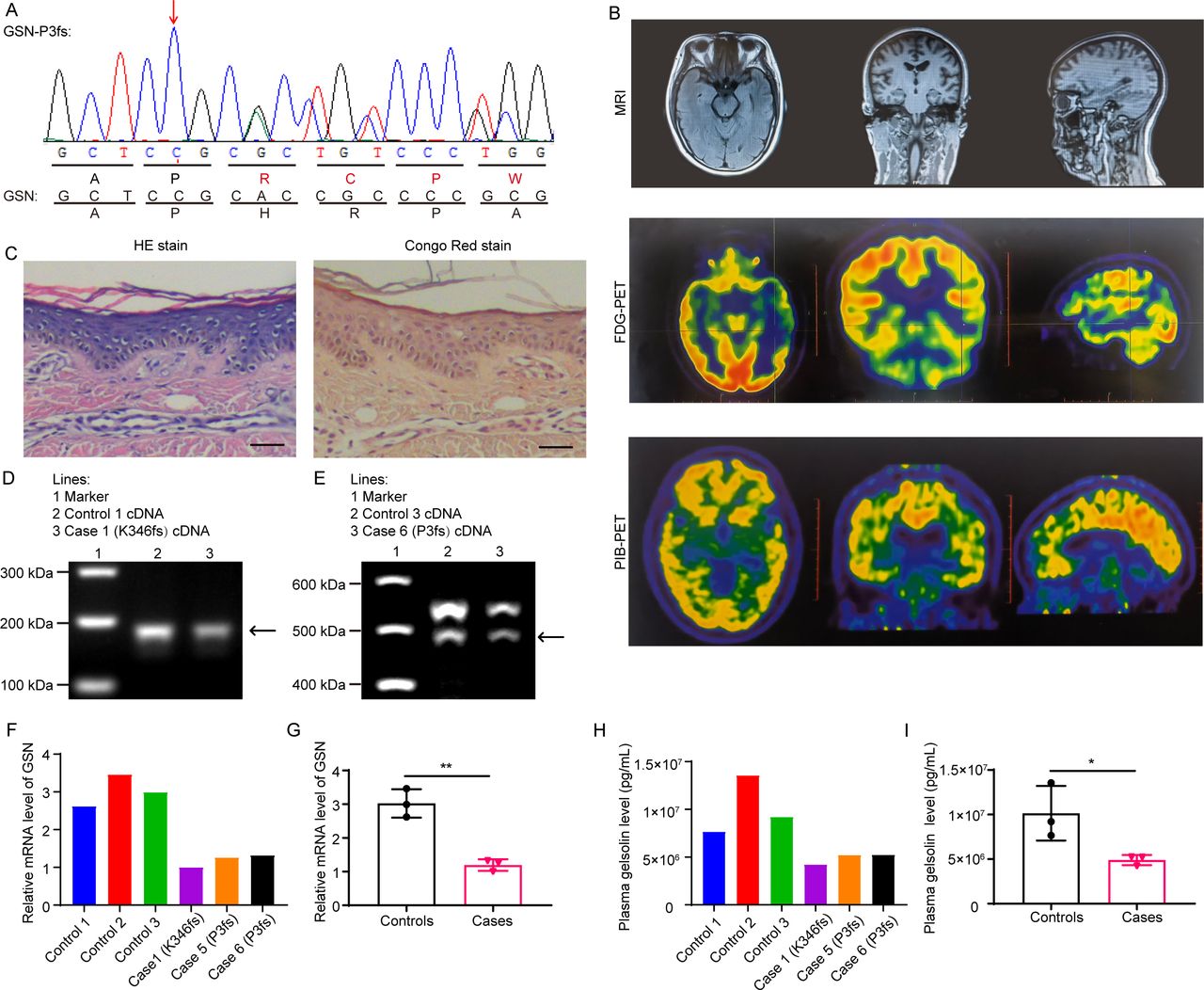 Figure 1
Figure 1 Examination results of case 6 and the reduced GSN expression in patients with Alzheimer’s disease with GSN frameshift mutations. (A) Nucleotide and amino acid sequences of case 6 (GSN:c.8_35del:p. P3fs) and GSN. (B) MRI, FDG-PET and PiB-PET of case 6 with P3fs mutation. (C) The H&E staining and Congo red staining of the skin biopsy showed no amyloid deposition of case 6 with P3fs mutation. Scale bar: 50 µm. (D) The same band between case 1 and control 1 in the electrophoresis of RT-PCR products. (E) The same band between case 6 and control 3 in the electrophoresis of RT-PCR products. (F) GSN mRNA expression level in case 1, cases 5 and 6, and controls 1–3 by qPCR. (G) Analysis of the data in (F). Reduced GSN mRNA expression level in cases compared with controls. n=3 per group. (H) Plasma GSN level in case 1, cases 5 and 6, and controls 1–3 by ELISA. (I) Analysis of the data in (H). Reduced plasma GSN level in cases compared with controls. n=3 per group. *P<0.05, **P<0.01. FDG-PET, 18F-fluorodeoxyglucose positron emission tomography; GSN, gelsolin; qPCR, quantitative PCR; RT-PCR, real-time PCR.
Plasma GSN detectionThe venous blood samples were collected using standard venipuncture protocols after an overnight fast. Then, plasma was collected in 30 min by centrifugation at 1000 rpm and 4℃ for 10 min. The plasma GSN level was detected by GSN ELISA kit (catalogue number ab270215; Abcam, USA). All procedures were performed in accordance with the manufacturer’s instructions.
CSF GSN and AD biomarker detectionThe CSF sample was collected according to international guidelines.15 Samples were centrifuged at 2000×g and 4℃ for 10 min and stored at −80°C. The CSF GSN level was detected by GSN ELISA kit (catalogue number ab270215, Abcam). All procedures were performed in accordance with the manufacturer’s instructions.
All CSF AD biomarkers (Aβ42, Aβ40, t-tau and p-tau) were measured using ELISA kits (catalogue numbers EQ 6511-9601, EQ 6521-9601, EQ 6591-9601, EQ 6531-9601; EUROIMMUN, Germany), and the detection was performed by experienced technicians in strict accordance with the instructions of the manufacturer within 1 week of sample collection.
Cell cultureThe SH-SY5Y cells (catalogue number ZQ0050; Zhong Qiao Xin Zhou Biotechnology, Shanghai, China) were cultured in high glucose Dulbecco's Modified Eagle's Medium (DMEM) (catalogue number C11995500BT, Gibco) supplemented with 10% Fetal Bovine Serum (FBS) and 1% PS at 37°C and 5% CO2 in a humidified atmosphere. The SH-SY5Y-APPswe stable cell lines were constructed by lentiviral infection with the APPswe mutation (specials: Homo sapiens, NM_201414.3: c.1785G>C p.K595N; c.1786A>C p.M595L).16 The control cell lines were transfected with empty lentivirus (EV). Western blot analysis of APP was performed to demonstrate the successful construction of APPswe cell lines (online supplemental figure S2).
 Figure 2
Figure 2 GSN frameshift mutations failed to reduce the Aβ level and apoptosis in SH-SY5Y-APPswe cells. (A) Immunofluorescence images of SH-SY5Y-APPswe cells transfected with GSN plasmids. SH-SY5Y-APPswe cells were characterised with GFP (green). SH-SY5Y cells transfected with GSN plasmids were characterised with DsRed (red). SH-SY5Y-APPswe cells transfected with GSN plasmids were characterised with both GFP (green) and DsRed (red), which appeared yellow in merged images. Scale bar: 20 µm. (B) Western blot analyses of the flag tag to show the expression of different GSN mutant proteins. GSN-WT was used as the negative control; GSN-D214N was the positive control (reported to be associated with FAF); GSN-K346fs and GSN-P3fs were frameshift mutations identified in patients with AD; GSN-R188I, GSN-Y301C and GSN-R525H were VUS identified in patients with AD and distributed in different domains of GSN (C) GSN K346fs mutation failed to reduce Aβ42 levels or the ratio of Aβ42/40 in SH-SY5Y-APPswe cells, as shown by ELISA analysis. n=3 per group. (D) ELISA analysis of Aβ42 and Aβ40 in SH-SY5Y-EV cells transfected with GSN plasmids. n=3 per group. (E) Immunostaining of Hoechst 33 342 (light blue) in SH-SY5Y-APPswe and SH-SY5Y-EV cells transfected with GSN plasmids. Scale bar: 50 µm. (F) Quantification of Hoechst-positive cells in (E). GSN K346fs and P3fs mutations failed to reduce apoptosis in SH-SY5Y-APPswe and SH-SY5Y-EV cells. n=6 per group. (G) ROS levels in SH-SY5Y-APPswe and SH-SY5Y-EV cells transfected with GSN plasmids. #P<0.05 vs the control group, *P<0.05 vs the WT group; #/*P<0.05, ##/**P<0.01, ###/***P<0.001, ####/****P<0.0001. Aβ, β-amyloid; AD, Alzheimer’s disease; DAPI, 4',6-diamidino-2'-phenylindole; DsRed: discosoma sp. red fluorescent protein; EV, empty lentivirus; FAF, familial amyloidosis of the Finnish type; GFP: green fluorescent protein; GSN, gelsolin; ROS, reactive oxygen species; VUS, variants of undetermined significance; WT, wild type.
Transfection of GSN plasmids and detection of the levels of Aβ42 and Aβ40A total of 5×104 cells (SH-SY5Y-APPswe and SH-SY5Y-EV) were sealed on a 24-well plate with 500 µL of medium. After 24 hours, cells were transfected with GSN plasmids using Lipofectamine 3000 Transfection Reagent (catalogue number L3000015, Thermo). The GSN plasmids with 3× flag tag were cloned into the eukaryotic expression pIRES2-DsRed2. The ratio of Lipo3000/DNA was 1.5 µL/µg. Six hours later, the culture medium was changed to DMEM with 10% FBS. Then, the cell culture was collected to detect Aβ levels by ELISA, and cells were calculated for apoptosis by Hoechst staining17 after 48 hours. The concentrations of Aβ42 and Aβ40 were measured using commercial ELISA kits (catalogue numbers DAB142 and DAB140B; R&D, USA). All procedures were performed in accordance with the manufacturer’s instructions.
Hoechst staining and apoptosis analysisCells were stained with Hoechst 33 342 (catalogue number 40 732ES03; Yeasen, China) after fixation according to the manufacturer’s protocol. A fluorescence microscope (Carl Zeiss Axio Imager 2; Carl Zeiss, Germany) was used to obtain the image. As the nuclei of apoptotic cells were concentrated, apoptotic cells were observed to have high fluorescence intensity by Hoechst staining and were noted as Hoechst-positive cells.17 The percentage of Hoechst-positive cells was analysed by ImageJ software.
ROS streaming detectionA total of 3×105 cells (SH-SY5Y-APPswe and SH-SY5Y-EV) were sealed on a six-well plate with 2 mL of medium. The same procedures were performed to transfect GSN plasmids as mentioned previously. The cells were collected for ROS streaming detection using a Cell Metre Fluorimetric Intracellular Total ROS Activity Assay Kit Brite 670 nm (catalogue number 22903; AAT Bioquest, USA). All procedures were performed in accordance with the manufacturer’s instruction.
CCK8 analysisA total of 5×104 SH-SY5Y cells were sealed on a 24-well plate with 500 µL of medium. After 24 hours, the culture media were changed to media supplemented with different concentration gradients of Aβ40 (0, 1, 5, 10 and 20 µM). The CCK8 Kit (catalogue number ab228554, Abcam) was applied to cells after 48 hours to evaluate the toxicity of the Aβ40 according to the manufacturer’s instructions.
Live/dead cell stainingThe SH-SY5Y cells were sealed on a 24-well plate with 500 µL of medium. Twenty-four hours later, the cells were transfected with GSN plasmids, and the procedures were the same as mentioned previously. After 6 hours, the culture medium was changed to DMEM with 10% FBS and 5 µM Aβ40. The treated cells were washed twice with assay buffer and stained with calcein-AM (4 µM) and PI solution (9 µM) using the Calcein-acetoxymethyl ester (AM)/propidium iodide (PI) Double Stain Kit (catalogue number 40 747ES76, Yeasen). After incubation for 30 min at 37°C, the cells were washed and photographed using a fluorescence microscope. The percentages of dead cells (calcein-AM–PI+) were calculated.18
Western blot analysesCells were collected and resuspended in RIPA lysis buffer (catalogue number P0013B; Beyotime, China) containing a protease and phosphatase inhibitor mixture. Cell suspensions were lysed on ice for 30 min, sonicated and centrifuged at 13 000 rpm for 10 min at 4°C. Briefly, protein concentrations were estimated using BCA Kit (catalogue number 70-PQ0012; MULTI SCIENCES, China). Lysates were separated on 10% or 12% sodium dodecyl sulfate–polyacrylamide electrophoresis gels (catalogue number P1200; Solarbio, China). After separation, the protein was transferred to a Poly Vinyli Dene Fluoride (PVDF) membrane (catalogue numbers IPVH00010 and ISEQ00010; Millipore, USA), and non-specific binding sites were blocked by treatmentwith commercial western blocking buffer (catalogue number SW3010, Solarbio) followed by antibody incubation: flag tag (1:1000, catalogue number A00187; GenScript, China), APP (1:1000, catalogue number ab32136; Abcam) and β-actin (1: 1000, catalogue number O10313; TransGen, China). Horse radish peroxidase (HRP)-conjugated secondary antibodies (1: 5000, Solarbio) were used. Western blot images were captured by the ChemiDocTM XRS+ with Image LabSoftware (Bio-Rad).
Statistical analysis
All measurement data are presented as mean±SD. Unpaired, two-tailed Student’s t-test was used to analyse the differences between two groups. Sex and APOE phenotype were analysed by χ2 test. Statistical analysis of multiple-group comparisons was performed by one-way analysis of variance. The correlation analyses were adopted Pearson or Spearman correlation analysis. The significance of gene enrichment was measured by Fisher’s exact test.19 GraphPad Prism software V.8.0 and SPSS V.23.0 were used for the aforementioned statistical analysis.
The details of the three-dimensional (3D) model structure and the scales of global cerebral atrophy–frontal subscale (GCA-F), posterior atrophy (PA) and medial temporal lobe atrophy (MTA) are described in the online supplemental methods.
DiscussionTo our knowledge, this study was the first to clarify the role of GSN frameshift mutations in the pathogenesis of AD, and proposed the genotype–phenotype heterogeneity of the GSN gene in which missense mutations lead to FAF, while frameshift mutations cause AD. Besides, the detection of higher plasma GSN levels in patients with AD at an early stage, indicating it possibly would be a potential diagnosis biomarker and monitoring the progression of AD (figure 5).
In this study, we first completed AD and FAF-related examinations in patients with GSN frameshift mutations, confirming that patients with GSN frameshift mutations were AD phenotype. However, none of the five patients with AD with GSN frameshift mutations, reported in the previous study,8 completed the exclusive FAF diagnosis.7 Therefore, our present study first demonstrates that frameshift mutations may cause AD phenotype.
Combined with our previous clinical cohort, the frameshift mutations (K346fs and P3fs) of GSN could account for 0.5% of AD cases. Moreover, frameshift mutations in the GSN gene in patients with AD were only observed in Han populations. Among the reported mutations in the GSN gene, there are three frameshift mutations (A34fs, K346fs and P3fs8 26), all of which were reported in Han populations, suggesting geographical heterogeneity of the GSN mutations. For missense variants, we failed to identify significant enrichment of these missense variants across the GSN gene in patients with AD compared with the controls. Interestingly, reported patients with FAF with GSN missense mutations were usually cognitive normal.7 27–30 Only one study reported that patients with FAF appeared slightly with abnormalities in visuocontructional and spatial performance,31 which further confirmed the genotype–phenotype heterogeneity of the GSN gene. The phenomenon of genotype–phenotype heterogeneity has also been reported in other genes previously. Wang et al 32 demonstrated that frameshift mutations of presenilin1 (PS1) led to familial acne inversa, which is a dermatological disease. However, pathogenic missense mutations of the PS1 gene were first reported to cause familial AD.33 No frameshift mutation was reported as a pathogenic mutation of the PS1 gene in AD. The different types of mutations of the PS1 gene contributed to completely different diseases. These studies suggest that different mutational forms of GSN lead to different clinical phenotypes and may be related to the pathogenesis of these diseases.
To figure out that frameshift mutations, rather than missense variants, are involved in AD pathogenesis, we first detected the mRNA and protein levels of GSN and found that the mutant mRNA could not be detected in patients, suggesting that the frameshift mutations did not generate fragments of GSN protein, which might be explained by nonsense-mediated mRNA decay.34 This was different from the pathogenic process of FAF. As the protein conformation changed in the acknowledged mutations of D214N or N211K,35 36 GSN could be cleaved when passing through the Golgi, and small fragments were generated during the process. The deposition of fragments on corresponding tissues led to the clinical symptoms of FAF.37–41 Nevertheless, no fragments were observed in patients with AD with GSN frameshift mutations at the mRNA or protein level. Additionally, we found patients with AD with GSN frameshift mutation had a decreased plasma GSN level, which was not present in patients with AD with identified missense mutations, indicating that the mechanism of loss of function might be involved in the pathogenesis of AD.
Mechanism studies found that GSN contained two Aβ binding sites and could bind and sequester Aβ,42 then inhibit Aβ fibrosis and degrade fibres that already formed.43 44 GSN could also inhibit Aβ-induced cytotoxicity by inhibiting apoptotic mitochondrial changes45 and protect neurons from apoptosis by actin remodelling.46 Then, we selected representative mutation sites and forms to clarify the mechanism of GSN frameshift mutations from the cytological level by observing the Aβ42 levels and cellular apoptosis in AD model cells transfected with different GSN plasmids. Meaningfully, compared with GSN-WT plasmid treatment, the increased level of Aβ42, the higher ratio of dead cells:live cells in the Aβ-induced toxicity tests, and more apoptotic cells were observed in the GSN-K346fs and GSN-P3fs plasmid-treated group, suggesting that the K346fs and P3fs mutations lost the protective function of GSN. For groups of missense mutations of GSN, no significant difference was observed, indicating that the mutant protein may retain its function to a certain extent. To our knowledge, the GSN protein can bind Aβ, inhibit its aggregation, and protect cells from apoptosis and ROS.4 47 Injection or overexpression of GSN resulted in a significant reduction in amyloid loads and a decrease in Aβ levels in AD transgenic mice.44 48 49 Our results showed that GSN frameshift mutations, rather than identified missense mutations, led to the GSN loss of function in reducing Aβ42 levels, inhibiting Aβ-induced toxicity, and protecting cells from apoptosis. This was different from the pathogenic process of missense mutations in FAF, which generate small fragments and deposit on tissues of eye, skin and peripheral nerves.37–41 Thus, different mutational forms of the same gene lead to different pathogenic pathways and present different clinical phenotypes.
Considering that GSN acts as an antiamyloidogenic protein and might change in AD progression,4 24 it may be a potential biomarker for AD. However, the plasma GSN levels were inconsistent in patients with AD reported in previous studies.22 24 In our cohort, the plasma GSN level was higher in early stage of disease duration of AD, and it was lower in medium and late stages of AD than that in controls, suggesting that GSN may function in the compensatory mechanism of the Aβ pathology. As the level of plasma GSN changes as AD progresses, the plasma GSN may be a novel potential biomarker for AD. To further explore the relationship between plasma and CSF GSN level, we also detected GSN level in CSF. The results showed a positive correlation between CSF GSN and plasma GSN, which increased the importance of plasma GSN as a biomarker. Additionally, the level of plasma GSN in patients with frameshift mutations was significantly decreased, while the level of plasma GSN in patients with missense mutations was comparable to that in controls, further confirming the loss-of-function mechanism of GSN frameshift mutations.
In summary, we found that GSN frameshift mutations are associated with the pathogenesis of AD. The level of GSN increases in the early stage of sporadic AD and has a protective effect, while in patients with frameshift mutations or patients with advanced disease, the level of GSN is downregulated and leads to the development or aggravation of the disease.
留言 (0)