The presence of pterin-containing molybdenum centers has been confirmed in more than 50 enzymes, and these enzymes are involved in the global cycling of nitrogen, sulfur, carbon, and arsenic [[1], [2], [3]]. These enzymes can be classified into three families based on, in part, the coordination sphere of the molybdenum center (Fig. 1). At the fully oxidized state, the Mo center is coordinated by at least one terminal oxo (e.g., in sulfite oxidase, DMSO reductase) or sulfido (e.g., in periplasmic nitrate reductase) group. In addition, Mo is coordinated by one or two pyranopterin (also called molybdopterin) cofactors through its dithiolate moiety. Thus, the Mo center is coordinated by multiple sulfur donors, e.g., in xanthine oxidase and sulfite oxidase, the Mo center is coordinated by three sulfur donors; in DMSO reductase, it is coordinated by four sulfur donors, while in periplasmic nitrate reductase, the Mo center is coordinated by at least five sulfur donors. However, in recent structural studies, the terminal oxo group has been replaced by a terminal sulfido group, suggesting that the Mo center is coordinated by six sulfur donors. Endogenous ligation to the Mo center is an interesting structural feature e.g., coordination by cysteine sulfur in sulfite oxidase and periplasmic nitrate reductase or coordination by serine oxygen in DMSO reductase and TMAO reductase. Structural studies on DMSO reductases have shown variations in the O(oxo)-Mo-O(ser) angle [4]. The endogenous ligation has been proposed to control the reactivity modulating the energetics of substrate binding [5,6]. Substrate transformation often involves a redox-driven (two-electron) oxygen atom transfer, with the catalytically competent state being regenerated via coupled proton-electron transfer processes [7]. During a catalytic cycle, molybdenum centers shuttle between +6, +5, and + 4 oxidation states [1]. Thus, the Mo(VI/V) and Mo(V/IV) reduction potentials play an important role in catalysis. Structural studies have been conducted on either the MoIV or the MoVI states; the structural information on the transient MoV state relies heavily on spectroscopy. We had suggested that the O(oxo)-Mo-O(ser) angular variation can modulate Mo(V/IV) reduction potential that can gate the electron transfer process, which we called ‘serine gated electron transfer’. [8,9] In this case, we used a heteroscorpionate molybdenum system that has shown to be useful for a better understanding of the catalytic processes [10,11]. Of the three Mo states, the +5 state is paramagnetic and is amenable to electron paramagnetic resonance (EPR) spectroscopy. In the past decades, several discrete inorganic complexes have been developed to understand the reactivity, structural and spectroscopic features in mononuclear molybdoenzymes [[12], [13], [14], [15], [16], [17], [18]]. Of particular interest here are the paramagnetic MoV complexes with different O(oxo)-Mo-O angles that allow a better understanding of the transient MoV in the enzymes. The EPR spin Hamiltonian parameters for MoV complexes and enzymes can be extracted from single-crystal or frozen solution data. Although electronic g-tensors, hyperfine A-tensors, and their orientations with respect to the molecular structure can be uniquely obtained from single-crystal data, preparation of magnetically diluted single crystals is not straightforward, and only a few such investigations for oxo-Mo(V) compounds have been reported [[19], [20], [21], [22]]. In addition, while the principal components of g and A can be determined from frozen solution EPR data, it is often impossible or at least difficult to find a unique solution for orientations of g and A with respect to the molecular structure from EPR simulation of data [23,24]. Fortunately, density functional theory (DFT) calculations can provide this missing link [[25], [26], [27], [28], [29], [30]].
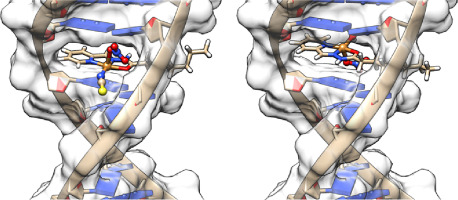
Calculation of a variety of spectroscopic properties of transition-metal complexes using ab initio and DFT has undergone rapid growth in recent years and has become a routine method in interpreting experimental findings [[31], [32], [33], [34]]. Although computationally more expensive ab initio techniques such as CASCSF, MRCI, CISD, and CCSD provide excellent agreements between theory and spectroscopic data, they can only be effectively applied to small molecule systems. A DFT approach, offers computational speed, allowing calculations of spectroscopic signatures of moderately large transition-metal complexes. More importantly, results obtained from DFT methods are comparable to low-level ab initio methods in accuracy. Indeed, DFT and TDDFT methods have been successfully used for calculations of UV–vis, CD, MCD, NMR, and Mössbauer spectra of numerous inorganic and organometallic compounds in recent years [[35], [36], [37], [38]]. We have also successfully applied DFT and TDDFT methods in calculating spectroscopic features (e.g., NMR [39], EPR [30] and UV–visible [8,[40], [41], [42], [43], [44], [45], [46]] in a variety of inorganic compounds including oxo‑molybdenum complexes. Until recently, however, calculating the EPR parameters using a DFT approach was quite challenging. Indeed, Neese, Kaupp and others have successfully implemented the treatment of relativistic effects for calculating EPR parameters of transition-metal complexes containing heavier metal ions [28,[47], [48], [49], [50], [51]]. Consequently, relatively few DFT-based studies have reported the calculation of g- and A-tensors and their orientations with respect to the molecular structure in paramagnetic MoV complexes. We have been interested in understanding the effect of geometric isomers on the ligand field and electronic structures in oxo‑molybdenum complexes [8,[52], [53], [54]]. Here, we show how geometric isomers of the same ligands affect the g- and A-tensors, and their orientations, in four pairs of the following cis- and trans-isomers: cis- and trans-(L1O)MoOCl2 complexes [L1OH = bis(3,5-dimethylpyrazolyl)-3-tert-butyl-2-hydroxy-5-methylphenyl)methane] cis,cis- and cis,trans-(L-N2S2)MoOCl [L-N2S2H2 = N,N′-dimethyl-N,N′-bis(mercaptophenyl)ethylenediamine], cis,cis- and cis,trans-(L-N2S2)MoO(SCN), and cis- and trans-(dt)2MoO(OMe) [dtH2 = 2,3-dimercapto-2-butene] [9,55]. Calculated EPR parameters are compared with the experimental values either reported in the literature (e.g., cis,trans-(L-N2S2)MoOCl, cis,cis- and cis,trans-(L-N2S2)MoO(SCN)) or determined by us (e.g., cis- and trans-(L1O)MoOCl2 complexes). For complete understanding, we also report the EPR parameters of a hypothetical system, i.e., cis- and trans-[(dt)2MoO(OMe)]2−. These complexes are shown in Fig. 2.
The cis- and trans-(L1O)MoVOCl2 complexes were prepared as described elsewhere [8], with the following modifications. Once the reaction between the L1OH ligand and MoCl5 was complete, as determined by TLC analysis, the resultant brownish-red solution was immersed in acetone-dry ice bath and cooled for 20–30 min. Next, hexane that was cooled similarly was added to induce precipitation. The resultant brown-red residue was filtered (yielded only a small amount that was not characterized), washed with ice-cold hexane, and the filtrate was evaporated to dryness, yielding a brown-red solid. This solid was dissolved in cold cholorform:acetonitrile (90:10 v/v) and flash chromatographed at 4 °C through an alumina column, with all resultant fractions placed in acetone - dry ice bath until tested for purity via TLC. Final purification was achieved by preparative TLC with chloroform as the eluent. Once the TLC was complete, the pink cis and green trans fractions were pooled into like fractions and evaporated to dryness. All samples for EPR spectroscopy were dissolved in a dry, degassed, 50:50 v/v toluene:chloroform mixture and immediately cooled to −77 °C until analyzed.
X-band EPR spectra were acquired using a Bruker-300 spectrometer with an Oxford ESR-910 liquid helium cryostat. The quantification of signals was relative to a spin standard (CuEDTA), and the spectra were obtained with a field modulation of 1 mTpp at 100 KHz. The magnetic field was calibrated with an NMR gaussmeter, and the microwave frequency was measured with a counter. The room temperature spectra of the two samples, shown in Fig. 3, Fig. 4, were recorded immediately after thawing the samples. After 40 min of incubation at room temperature, the cis-isomer isomerizes to the trans-isomer (∼40%), which can be differentiated by EPR spectra and is consistent with the previously reported results.
The EPR spectra were simulated with the standard spin Hamiltonian in Eq. (1),H=βB•g•S+hS•A•Iwhere the principal axes of the g-tensor may not be coincident with the principal axes of the A-tensor. The program calculates spectra with the proper ratio of nuclear species 96/98/100Mo (I = 0) to 95/97Mo (I = 5/2), 74.5:25.5. Three Euler angles (αxy, βxz, and γyz) were included to rotate the A tensor relative to the g tensor. The isotropic g and A values were first determined from simulations of the room temperature spectra of the two samples. The low-temperature spectra were then fitted least-squares by allowing the g-values, A-values, and angles to vary. The automated fitting was constrained so that the isotropic values of the g and A tensors matched the values obtained from the room temperature spectra. In addition, the symmetry of the complexes renders γyz meaningless. Thus, the number of independent variables in the fitting procedure was reduced from 9 to 6, and the simulations determined a unique parameter set.
Geometries of all MoV complexes discussed have been optimized using unrestricted DFT formalism implemented in the Gaussian 09 program [56]. In each case, the complete molecular structures have been used in geometry optimization jobs without restrictions. In order to investigate the influence of the exchange-correlation functional and the amount of the Hartree-Fock exchange on the calculated geometries and predicted spin Hamiltonian parameters, the following three functional were used: (i) Becke's 1988 GGA exchange functional (0% of Hartree-Fock exchange) coupled with the Perdew's 1981 local correlation functional (BP86) [57,58]; (ii) Becke's three-parameter hybrid (∼20% of Hartree-Fock exchange) functional [59] along with the Perdew non-local correlation functional (B3P86) [60]; (iii) 1996 pure functional of Perdew, Burke, and Ernzerhof, as made into a hybrid (25% of Hartree-Fock exchange) by Adamo (PBE0, also denoted in Gaussian as PBE1PBE) [61]. In all geometry optimizations, full electron DZVP and 6-311G(d) basis sets were used for molybdenum, and all other atoms, respectively [62,63]. To ensure that optimized geometries represent minima on potential energy surface, frequencies were calculated for all optimized geometries.
All spin Hamiltonian parameters were calculated using ORCA 2.6, 2.7, and 2.8 programs [64]. Zero-order regular approximation (ZORA) was used at the all-electron level to treat the scalar relativistic effects. A tight convergence criterion was used for all spin-unrestricted DFT calculations using BP86, B3P86, and PBE0 exchange-correlation functional mentioned in geometry optimization section. Triple ζ quality polarized TZVP basis set was used for all atoms in EPR calculations. Implemented into ORCA, split-RI-J Coulomb approximation was used in BP86 calculations using appropriate TZVP auxiliary basis. The basis set on the molybdenum atom was completely decontracted to add flexibility and to improve the calculation of the Fermi contact term. Increased grid quality and integration accuracy was also applied to all atoms in the first coordination sphere of the molybdenum atom. The g matrix was calculated (as implemented in the ORCA program) as a sum of four contributions (eq. 2):g=ge+ΔgRMC+ΔgDC+ΔgOZ/SOC
The first term equals the g value of the free electron (ge = 2.002319) and isotropic in nature. The second term represents a relativistic mass correction, which can be calculated based on the kinetic energy integrals and the ground state spin density as:ΔgRMC=−α2S∑ijPijα−β<ϕi∣Τ∣ϕj>
Here (Eq. (3)) α is the fine structure constant, the total spin of the ground state is represented by S, Pijα-β is the SCF-based unrestricted spin density matrix, φ represents the Gaussian basic set, and T is the kinetic energy operator (T=−1/2∇2). The third term in eq. 2 is a diamagnetic correction term that can be calculated using SCF spin density as (eq. 4).ΔgDC=12S∑ijPijα−β<ϕi∣∑AξrAr→Ar→−r→A,rr→∣ϕj>
In Eq. (4), r→A represents the position vector of the electron with respect to nucleus A, while r→ the position vector relative to the gauge origin, and ξrA is a function, which can be expressed as in eq. 5.ξriA=α22ZeffAri−RA3
Here, ZeffA is a semi-empirically estimated effective nuclear charge of atom A at position RA. The operator described in eq. 5 has been parameterized by Koseki and gives good results for transition-metal compounds. The final term in eq. 2, (ΔgOZ/SOC), which is the major perturbation to overall Δg, arises as a cross term between the orbital Zeeman (this can be estimated using gauge including atomic orbital, GIAO method) (Eq. (6)) and spin-orbit coupling (eq. 7) operators and can be calculated using ORCA following Eq. (8):HOZ=β∑iBliHSOC=∑iξriAlAisiΔgOZ/SOC=1βS∂2E∂Br∂μswhere l(i) is the angular momentum operator of the ith electron relative to the chosen gauge origin, s(i) is a spin operator for the ith electron, lA(i) is the angular momentum operator of the ith electron relative to nucleus A, and μs is the magnetic moment of the electron. Implementation of eq. 2 into coupled perturbed SCF theory both at Hartree-Fock and Density Functional Theory levels has been described in the literature.
The principal values of the hyperfine coupling AMo tensor were calculated as the sum of isotropic Fermi contact term, anisotropic spin dipolar contribution, and spin-orbit coupling contribution [64]. The percent contribution that atomic orbitals lend to their respective molecular orbitals was calculated using the VMOdes program [65].




留言 (0)