Study design
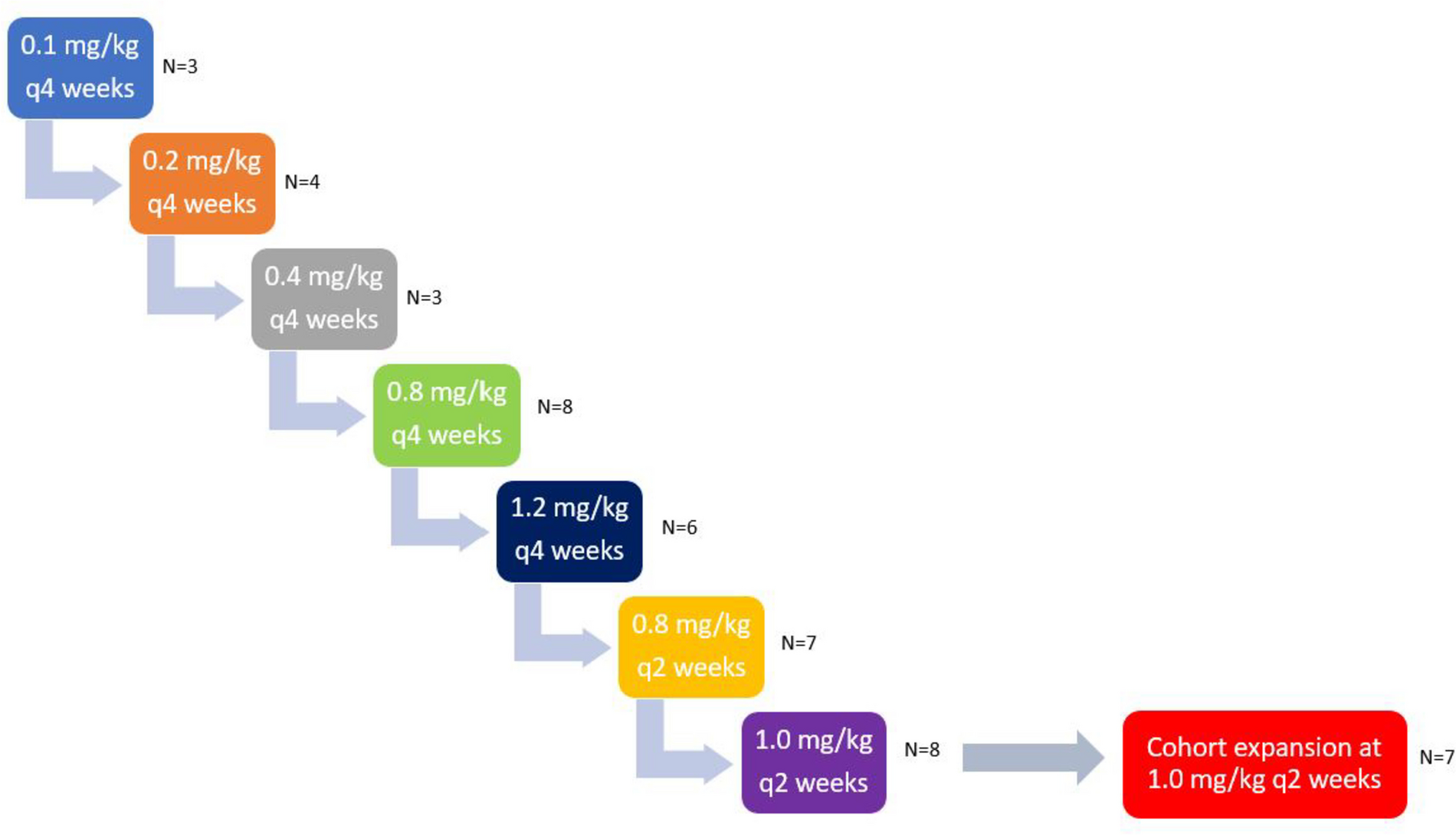
This was a Phase Ib, multicenter, open-label, dose-finding clinical trial (NCT02610075). The primary objective was to determine the MTD and RP2D of oral adavosertib monotherapy administered qd or bid in patients with locally advanced or metastatic solid tumors. Various doses of adavosertib were assessed: bid 1 (125 mg), bid 2 (150 mg), qd 1 (200 mg), qd 2 (250 mg) and qd 3 (300 mg). Three types of treatment schedules were investigated: cohorts bid 1, bid 2, qd 1.1, qd 2.1 and qd 3.1 received bid or qd treatment on a 5/9 schedule (5 days on treatment, followed by 9 days off) on a 14-day cycle. Cohorts qd 1.2, qd 2.2, and qd 3.2 received treatment on a 5/2 schedule (5 days on followed by 2 days off for 2 of 3 weeks) in a 21-day cycle. Cohorts qd 2.3 and qd 3.3 received treatment on a weekly 5/2 schedule in a 21-day cycle (Table 1). The highest dose level(s) at which less than one-third of evaluable patients (none of 3 patients or 1 of 6 patients) experienced a dose-limiting toxicity (DLT) was declared the MTD.
Table 1 Baseline patient demographics and characteristics
The secondary objectives of this study were to evaluate the safety and tolerability, preliminary antitumor activity and pharmacokinetics (PK) of adavosertib monotherapy.
Patients
Eligible patients were ≥ 18 years of age with an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1 and measurable or non-measurable disease (Response Evaluation Criteria in Solid Tumors [RECIST] v1.1) [17]. Histological or cytological evidence of locally advanced or metastatic solid tumors (excluding lymphoma), for which standard therapy did not exist or had proven ineffective or intolerable, was required.
Exclusion criteria included, but were not limited to: use of anticancer drugs within 21 days or 5 half-lives, whichever was shorter, prior to the first adavosertib dose, with a minimum of 10 days between termination of the prior treatment and administration of adavosertib treatment.
Antiemetic prophylaxis was mandatory. Prior to each adavosertib dose, patients received an oral serotonin 5-HT3 antagonist: ondansetron 8 mg bid or granisetron 1 mg bid. For patients receiving the qd dosing schedule, a second dose of antiemetics could be administered 8 h later if nausea and vomiting continued. Oral dexamethasone 4 mg was given on day 1 of each adavosertib dosing period, unless contraindicated or not well tolerated. Aprepitant and fosaprepitant were not permitted because of known drug–drug interactions.
Patients could continue adavosertib treatment until disease progression, unacceptable toxicity, or any other discontinuation criterion was met.
An independent ethics committee/institutional review board approved the final clinical study protocol, including the final version of the informed consent form and any other written information provided to participants. All participants provided written informed consent, and the study was conducted in accordance with the Declaration of Helsinki, the International Council for Harmonisation, good clinical practice, applicable regulatory requirements, and the AstraZeneca policy on bioethics [18].
Safety
Safety data were recorded throughout the study until 30 days after the last adavosertib treatment, using the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) v4.03.
DLTs were evaluated during the first 28 days (5/9 schedule) or the first 21 days (5/2 schedule) of treatment. Patients who received less than 80% of the planned adavosertib dose for the treatment cycle were not included in the analysis unless it was due to a DLT. DLTs were defined as toxicities related to adavosertib treatment that met at least one of the following criteria: hematologic toxicities, such as neutropenia or thrombocytopenia (grade ≥ 4 for at least 7 days, including infection with febrile neutropenia); neutropenic fever (grade ≥ 3); thrombocytopenia (grade ≥ 3) with bleeding (grade ≥ 2); non-hematologic toxicity (grade ≥ 3); liver function tests – grade ≥ 3 total bilirubin, alanine aminotransferase or aspartate aminotransferase, or alkaline phosphatase lasting for > 48 h, or any change in liver function tests consistent with Hy’s Law; and any other clinically significant and/or unacceptable toxicity that did not respond to supportive care, resulted in a disruption of the dosing schedule of more than 7 days, or was judged to be a DLT by the investigator in collaboration with the medical monitor.
Efficacy
Preliminary efficacy was assessed using objective response rate (ORR) in patients with measurable disease, disease control rate (DCR) and progression-free survival (PFS), all in accordance with RECIST v1.1 [17]. Responses for ORR required confirmation after at least 4 weeks.
After initiation of treatment, tumor burden was assessed at baseline and every 8 weeks (± 1 week) for the qd 5/9 dosing, and every 9 weeks (± 1 week) for the qd 5/2 treatment schedule, respectively, using computed tomography or magnetic resonance imaging of the chest and abdomen or pelvis.
Archived tumor tissue of patients who consented and for whom a valid test result was obtained (n = 28) was analyzed using the Foundation Medicine (FMI) next-generation sequencing (NGS) platform to assess correlations between genomic profile and clinical outcomes [19].
Pharmacokinetics
Pharmacokinetic (non-food effect) analyses were performed on blood samples, which were collected pre-dose during treatment cycle 1 on days 1 and 5, and 1, 2, 4, 6, 8, 10 and 24 h after receiving the first adavosertib dose on days 1 and 5. Pre-dose samples were also collected on day 5 of treatment cycle 5.
The area under the plasma concentration–time curve from time 0 to 10 h post-dose (AUC0–10) and maximum concentration (Cmax) were determined.
Dose proportionality (non-food effect) of adavosertib AUC0–10 and Cmax for cycle 1, day 1 was assessed across the 125–300 mg dose range using the power model with the linear regression independently incorporating log-transformed AUC0–10 and Cmax. The slope estimate (β) and corresponding 90% confidence interval (CI) were calculated, where a β estimate of 1 with 90% CI entirely within the bounds of 0.8–1.25 indicated perfect dose proportionality.
For the preliminary food-effect analysis, blood samples were collected pre-dose and 1, 2, 4, 6, 8 and 10 h after receiving the first adavosertib dose during treatment cycle 1 on day 1 (fasted condition), and during treatment cycle 2 on day 1 (fed condition). Analyses were performed for the bid dosing schedules and RP2D only.
For the fasted condition, patients were required to fast for at least 10 h prior to receiving adavosertib until 4 h post-dose. Patients were allowed glucose and/or juice if they had signs or symptoms of hypoglycemia after receiving adavosertib. Water was restricted from 1 h pre-dose until 1 h post-dose, except for the 240 mL of water administered with treatment.
For the fed condition, patients fasted for at least 10 h prior to receiving adavosertib until 4 h post-dose, with the exception of a high-fat meal eaten in the 30 min before adavosertib administration. If the meal was not completed within 30 min, adavosertib was administered so long as 75% of the meal had been consumed within 45 min of starting the meal. The study was conducted using the Food and Drug Administration standard high-fat meal under fed conditions, which should have a total of 800 to 1000 kcal, with approximately 50% of the caloric content made up from fat [20].
Natural log-transformed AUC and Cmax were compared between fed/fasted conditions using a mixed effects analysis of variance model, which was fitted separately for each assessed dose. Estimates of the mean difference between treatments and corresponding 90% CI were calculated using a linear mixed effects model with a fixed effect for treatment (fed versus fasted) and a random effect for patients. Back transformed geometric means and 95% CIs were estimated for each condition.
Statistical methods
Statistical analyses were performed using SAS by Sarah Cannon Development Innovations, LLC under the direction of the AstraZeneca Biometrics Group. Descriptive statistics were used to summarize the safety, PK and preliminary antitumor activity data by treatment cohort.
Patients who received at least one dose of adavosertib were included in the full analysis set, which was used for safety and efficacy evaluations. The per protocol analysis set was defined as a subset of the full analysis set, excluding subjects having an important protocol deviation, to be used for analyses of efficacy endpoints if appropriate. There were no important protocol deviations; therefore, the patient groups included in the safety and efficacy evaluations were the same.
All patients who received at least one adavosertib dose and provided at least one PK sample were included in the PK analysis.

留言 (0)