
記住我
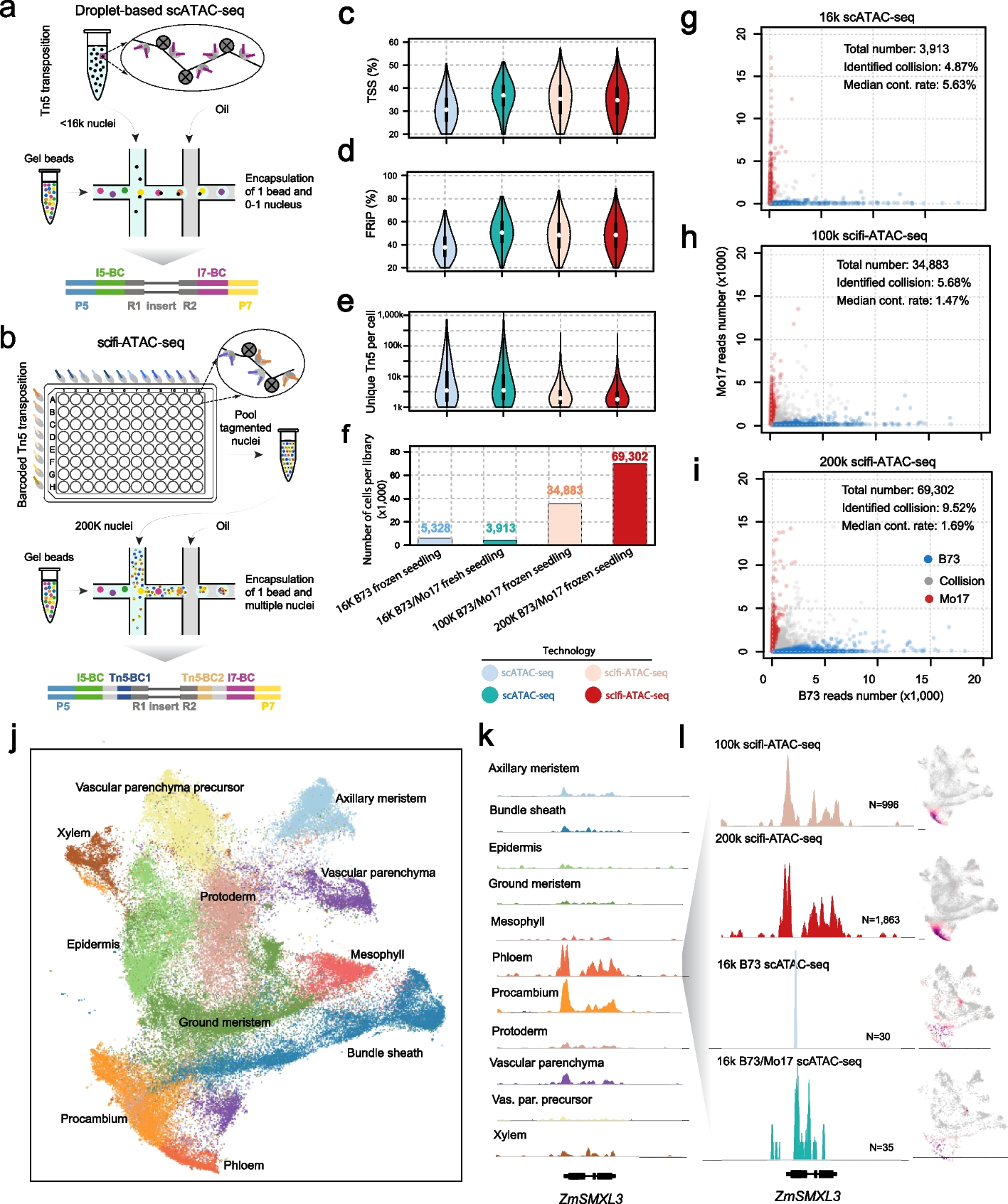
To study whether group variances are equal or unequal in scRNA-seq pseudo-bulk data, we explored pseudo-bulk scRNA-seq datasets generated with cells from specific cell types obtained from various sample types ranging from experimental replicates of mice to human samples (see the “Methods” section). We examined three things: (1) multi-dimensional scaling (MDS) plots, (2) common biological coefficient of variation (BCV) of groups, and (3) mean-variance trends derived from individual groups (we refer to these as “group-specific voom trends”). Larger distances between samples in a group on the MDS plots indicate more within-group variation. BCV is a measure of the biological variability in gene expression between biological replicates and is frequently used to estimate the variance of gene expression in RNA-seq data [29]. Higher common BCV values correspond to increased biological variation between samples across genes based on the assumption of the negative binomial (NB) distribution in edgeR. For the group-specific voom trends, we are interested in observing where the curves sit relative to other groups in the same study, as well as the shape of the curve. The shape and “height” of the curves reflect the total variation within groups—both technical and biological.
In studies of mouse lung tissue [30] and Xenopus tail [31], we observed some minor differences in group-specific voom trends, with the curves sitting close together and mostly overlapping one another (Fig. 1a-b). Common BCV values for these studies ranged from 0.197 to 0.240 across 2 groups in mouse lungs, and 0.226 to 0.295 across 5 groups in Xenopus tails.
Fig. 1
Group variation in scRNA-seq pseudo-bulk datasets. Group variation in 4 publicly available scRNA-seq datasets with various experimental designs, with replicates samples from a mouse lungs, b Xenopus tails, c human PBMCs, and d human lungs, is summarized. For each scRNA-seq dataset, cells of one particular type were selected (see the “Methods” section), and the cells from each sample were aggregated to create pseudo-bulk counts. Multidimensional scaling plots of pseudo-bulk data were plotted in the left panel, with distances computed from the log-CPM values and samples colored by groups. Group-wise common BCVs are plotted in the middle panel. Group-wise mean-variance trends are plotted in the right panel. Colors denote groups
In human datasets, the differences in group variances were greater. For human peripheral blood mononuclear cells (PBMCs) [32], healthy controls unsurprisingly exhibited lower variability than the 3 other patient groups to which they were compared (Fig. 1c). This was evident from group-specific voom trends—although the curves had similar shapes, the curve of the healthy controls sat distinctly below the curves of other groups. Common BCV values for the PBMCs ranged from 0.154 to 0.241, with the lowest for healthy controls and the highest for asymptomatic patients.
A separate study on human macrophages collected from lung tissues [33] showed even higher levels of heteroscedasticity, where common BCV values ranged from 0.338 to 0.495 (Fig. 1d). Group-specific voom trends had distinct shapes and were well separated from one another along the vertical axis (with the exception of IPF and control groups which were quite similar). Moreover, the plateauing of voom trends at higher expression values that are commonly observed in many datasets was not observed here. This might be on account of the complexity of regions in the lung where samples are collected, the diverse causes of lung fibrosis, and limited patient numbers for some groups. High levels of biological variation are reflected in the large BCV values in this dataset.
While we have not commented specifically on MDS plots, these plots (or other similar plots e.g., principle components analysis) provide a useful first glance of the data and are already part of many analysis pipelines (Fig. 1). For example, in the study of mouse lung tissue, the 3-month (3m) samples are less spread out across dimensions 1 and 2 than the 24-month (24m) samples, indicating that the 3m group has lower variability than the 24m group. This is confirmed by the groups’ BCV values and voom trends.
In conclusion, we observe unequal group variability across multiple scRNA-seq pseudo-bulk datasets. At this stage, it is unclear whether gold-standard bulk DE analysis methods are robust against heteroscedasticity, and how different group variances need to be before it affects their performance. We test this in later sections of this article, using three gold-standard methods that do not account for heteroscedasticity and two novel methods that do.
Novel use of voomWithQualityWeights using a block design (voomQWB)The first method that accounts for group-level variability makes novel use of the existing voomQW method. The standard use of voomQW assigns a different quality weight to each sample, which then adjusts the sample’s variance estimate—a strategy used to tackle individual outliers in the dataset. Rather than adjusting the variance of individual samples, we adjust the variance of whole groups by specifying sample group information via the var.group argument in the voomWithQualityWeights function. This produces quality weights as “blocks” within groups (identical weights for samples in the same group) and adjusts each group’s variance estimate—we refer to this method as “voomQW using a block design,” or simply voomQWB.
Figure 2a shows the estimated group-specific weights from voomQWB for a study comparing healthy controls to COVID-19 patients that are moderately sick and those that are asymptomatic [32]. Samples of moderately sick and asymptomatic patients have similar weights, just under 1; while the weights for healthy controls are above 1 (1.27). The sample weights are combined with observation-level weights derived from the overall mean-variance trend from voom (Fig. 2b). What this achieves in practice is an up-shift of the voom trend for groups with sample weights below 1 (Fig. 2c pink and green curves), resulting in a higher variance estimate and a smaller precision weight for statistical modeling (see the “Methods” section). On the other hand, groups with sample weights that are greater than 1 have a down-shifted voom trend (purple curve), resulting in lower variance estimates and larger precision weights. There are a couple of things to note here: (1) the group-specific voom trends from voomQWB (Fig. 1c) are roughly parallel to the single voom trend (Fig. 1b), and 2) the group-specific trends shown here are created manually, not as an output of the voomWithQualityWeights function.
Fig. 2
An overview of the voom-based mean-variance modeling methods applied. On the PBMC1 pseudo-bulk data, group-specific weights estimated using voomQWB by defining each block (group) as different levels of the symptom variable are plotted in (a). The equal weight (\(=1\)) level is plotted as a dashed line. Across all observations, gene-wise square-root residual standard deviations are plotted against average log-counts in gray in (b). voom applies a LOWESS trend (black curve) to capture the relationship between the gene-wise means and variances. Based on the final precision weights used in voomQWB, adjusted curves for each block are plotted in (c), where replicates in the same group share the same curve. Different colors and line types represent different groups (blocks). Dashed lines were used to avoid over-plotting. When voomByGroup is used, LOWESS trends are fitted separately to the data from individual groups to capture any distinct mean-variance trends that may be present (d–f). All group-specific trends from this dataset are plotted together in panel g, with different colors per group
voomByGroup: modeling observation-level variance in individual groupsAs mentioned above, voomQWB models group-wise mean-variance relationships via roughly parallel trend-lines, which has the disadvantage of not being able to capture more complicated shapes observed in different datasets (Fig. 1). The second method we describe here, called voomByGroup, can account for such group-level variability with greater flexibility. voomByGroup achieves this by subsetting the data and estimating separate voom trends for each group. In other words, while voomQWB can shift the same voom trend up and down for each group, voomByGroup estimates distinct group-specific trends that can also allow up- and down-shifts for different groups.
For example, on the PBMC1 dataset, the mean and variance are calculated for the log2counts-per-million (log-CPM) of each gene in “Group 1 (Moderate symptoms)” and a curve, or trend, is fitted to these values from which precision weights are then calculated (Fig. 2d). Similarly, a curve is fitted separately to each of the other groups in the dataset (Fig. 2e and f). This results in 3 non-parallel curves as shown in the summary plot (Fig. 2g) which includes all 3 trends. In theory, the voomByGroup method gives more robust estimates of variability since the trend for each group can take on a different “shape”—we test how this works in practice in the following sections.
Group variance methods provide a balance between power and error controlUsing simulated data, we test the performance of 3 gold-standard methods against the 2 new methods that account for heteroscedastic groups. The gold-standard methods are voom, edgeR using a likelihood-ratio test (edgeR LRT), and edgeR quasi-likelihood (edgeR QL). The methods that account for group heteroscedasticity (“group variance methods”) are voomQWB and voomByGroup. Using simulations of pseudo-bulk data, we can examine the effects of unequal group variation while controlling other factors. Specifically, group variation can be divided into biological variation between RNA samples and technical variation caused by sequencing technologies.
In the first scenario (scenario 1), we looked into unequal group variation as a result of biological variation. To obtain pseudo-bulk data, we simulated single-cell gene-wise read counts that followed a correlated negative binomial distribution and aggregated the reads from each sample (see the “Methods” section and Additional file 1: Fig. S1). Each simulation consists of 4 groups with 3 samples in each group—a total of 50 such simulations were generated. We generated varying group-specific common BCVs for the 4 groups that are well within the range of BCV values observed in experimental datasets (Figs. 1 and 3a)—the BCV values averaged over 50 simulations were 0.2, 0.22, 0.26, and 0.28 (the values in Fig. 3a are for one such simulation).
Fig. 3
Group variance modeling methods provide good power while controlling the false discovery rate. a Mean-variance trends plotted for each group by the voomByGroup function on simulated scRNA-seq data with varying group-specific common BCV values, where colors represent different groups. In terms of the simulated variability-level, groups 1 and 2 represent Low variation, while 3 and 4 have High variation. Based on DE analysis results, FDR across methods in different comparisons (colors denote the 3 comparison types) are summarized in panel (b) at a cut-off of 0.05. The number of DE genes recovered by different methods for comparison between the two groups with higher variability (panel c) and the two groups with lower variability (panel d) at the same FDR cut-off are shown. For each bar in these plots, gray represents true positive genes, red represents false positive genes, and the FDR is labeled at the top of the bar
The mean-variance trends generated for the different groups appear as expected, with a typical decreasing “voom-trend” with increasing gene expression and the curves ordered correctly from those with the most biological variation at the top of the plot (group 4 in Fig. 3a) to the group with the least biological variation at the bottom of the plot (group 1). The left-hand side of these mean-variance trends is primarily driven by technical variation—as expected, here they mostly overlap each other since groups were generated to have the same technical variation in these simulations. These group-specific mean-variance plots generated by the voomByGroup function provide a useful “first glance” of the data before any formal testing was carried out.
We then performed differential gene expression analysis for all pairwise group comparisons, which gave a total of 6 comparisons. In these simulations, 50 genes were generated to be upregulated in each group, such that 100 genes are differentially expressed in each pairwise comparison (see the “Methods” section). We noticed that the number of differentially expressed genes varied from method to method and calculated the false discovery rate (FDR) of each method which was averaged over the 50 simulations. The FDR, or type I error rate, is calculated as the number of genes that were incorrectly identified as differentially expressed out of the total number of genes that were identified as differentially expressed at a particular adjusted p-value cut-off. We observed that none of the methods controlled type I error for all comparisons across the 3 cut-offs we examined: adjusted p-value cut-off of 0.01, 0.05, and 0.10 (Fig. 3b, Additional file 1: Fig. S2); such that the methods detected false discoveries at a higher rate than expected. voomByGroup out-performed other methods by controlling type I error at the 0.01 and 0.10 cut-offs, and only exceeding the threshold marginally for 2 out of 6 comparisons at the 0.05 cut-off (FDR of 0.054 and 0.056).
A closer look at these plots revealed that the gold-standard methods had FDR values that spanned a broad range, with some comparisons having FDR values that were well under the threshold, and others that exceeded the threshold by 2- or 3-fold. This means that it could be difficult to gauge whether the DE results are too conservative, too liberal, or perhaps “just right” for a given comparison in real datasets when applying these methods to heteroscedastic groups. The range of FDR values is broadest for edgeR LRT, followed by edgeR QL, then voom. In comparison, the group variance methods, though not perfect in terms of type I error control, had a substantially tighter range of FDR values, and the comparisons that exceeded the FDR threshold only exceed it by a small margin.
To understand how heteroscedasticity influences DE analysis in more detail, we focused on results obtained using a 0.05 adjusted p-value cut-off. Across the 6 comparisons, group variance methods tend to detect similar numbers of differentially expressed genes, the same goes for gold-standard methods (Fig. 3c-d, Additional file 1: Fig. S3). There is some variation between the gold-standard and group variance modeling methods, with some comparisons having quite similar results, while others produce results that are very different. A closer examination of the comparison between group 3 and group 4 (which we refer to as “High vs High” in terms of biological variation) and group 1 and group 2 (“Low vs Low”) shows where the 2 classes of methods differ.
In the High vs High comparison, gold-standard methods detect more DE genes than group variance methods. However, the DE genes contain a much higher proportion of false discoveries than it was controlled for. It is not as though gold-standard methods were prioritizing false-positive genes in terms of significance—it had similar numbers of true- and false-positive genes when looking at top-ranked genes (Additional file 1: Fig. S4). Rather, gold-standard methods had smaller adjusted p-values than group variance methods, allowing more genes detected at a certain cut-off (Additional file 1: Fig. S5a). By pooling variance estimates across all 4 groups, gold-standard methods under-estimate the variances for groups 3 and 4, when in fact those groups have relatively high biological variation, resulting in poor type I error control. voomByGroup and voomQWB are more robust in their estimation of individual group variances, allowing them to maintain good type I error control.
In the Low vs Low comparison, all methods have good type I error control, with group variance methods detecting substantially more DE genes than gold-standard methods. Although top-ranked genes were yet again very similar (Additional file 1: Fig. S4), this time, gold-standard methods had larger adjusted p-values than group variance methods (Additional file 1: Fig. S5b), meaning that fewer genes were selected at given threshold. Here, the pooled variance estimates used by gold-standard methods resulted in an overestimation of variances for the two groups with relatively low biological variation (groups 1 and 2). In consequence, gold-standard methods suffered from a loss of power.
In the Low vs High comparison, there were no significant benefits in applying the group variance methods even though these methods provide more accurate variance estimates than the standard methods. When using voom, the overall variance trend that is applied to all samples and groups would under-estimate the variability of high variance groups, and over-estimate this for low variance groups. When pairwise comparisons are made between High and Low groups, the over- and under-estimated values balance each other out, giving results that are similar to the more precise estimates from voomByGroup and voomQWB.
These simulations demonstrated that in the presence of group heteroscedasticity, group variance methods have a good balance between controlling type I error and the power to detect DE genes. To ensure that the superior performance of group variance methods was due to group heteroscedasticity in the data, we separately simulated 50 null simulations (scenario 2) where all groups had equal underlying biological variation (see the “Methods” section). We observed similar numbers of true positives and false discoveries between gold-standard and group variance methods (Additional file 1: Fig. S6).
voomByGroup captures both biological and technical variation well in individual groupsSystematic differences are commonly observed in the sequenced libraries of scRNA-seq data [34]. For example, gene counts vary between cells on account of limited starting material per cell and variations in technical efficiency. In addition, the number of cells detected in each sample is also variable. After aggregating cells, pseudo-bulk samples have library sizes that are more variable than that of bulk RNA-seq data—contributing to a major source of technical variation in pseudo-bulk data.
We explore the influence of unequal library sizes by varying the number of cells in each sample for a new set of simulations. Keeping the underlying biological variation constant between groups (homoscedasticity), we first vary the library sizes of samples. The provided number of cells are 250, 250, and 250 in group 1, 250, 200, and 200 in group 2, 250, 500, and 500 in group 3, and 250, 750, and 750 in group 4 (Fig. 4a). The expected library size for each cell remains constant (see the “Methods” section).
Fig. 4
voomByGroup captures both biological and technical variation. a Summary of the simulation design with unequal numbers of cells per sample, with colors denoting the different groups in the dataset. In the scenario with technical variation only (unequal library sizes) across groups, mean-variance trends estimated by voomByGroup are plotted in panel (b), with group-wise common BCVs displayed in the top-right corner. Based on DE analysis results, FDRs across methods for different comparisons, denoted by distinct colors, are summarized in panel (c) at an FDR cut-off of 0.05
Under this scenario (scenario 3), the mean-variance trends generated appear to mostly overlap each other on the right-hand side as expected on account of equal group dispersions, while on the left-hand side, slight differences appear (Fig. 4b). DE analysis was then performed over 50 simulations and averaged FDR rates and numbers of DE genes were compared. We observed much tighter ranges of FDR values compared to those from scenario 1 (Fig. 4c, where the same y-axis range from Fig. 3b was used) and a similar number of true positive genes compared to the null simulation (Additional file 1: Fig. S7). These suggest that simulated technical variation does not have a significant influence on the DE results. To compare the influence of technical and biological variation in the simulations, we carried out another separate 50 simulations (scenario 4) with both aspects of variation incorporated (see the “Methods” section). For these results, we observed expected location trends that differed on both the left and right sides (Additional file 1: Fig. S8a). FDR results were rather similar to what was observed in the scenario where biological variation was unequal (Additional file 1: Fig. S8b, Fig. 3b), indicating that biological variation is the major source of variation that influences the DE results.
However, closer inspection of the FDR plot from scenario 3 where only the number of cells differed between groups revealed that among those comparisons, voom and voomQWB performed similarly, as a result of the weighting strategy used in voomQWB only adjusting the group-wise weight in an overall manner. While voomByGroup is more flexible, we observed that for group-wise mean-variance trends, regardless of the overlapping trends on the right-hand side, on the left-hand side, groups with fewer cells (group1 and group2) exhibit slightly more variation and sit at the top, while the group with the largest number of cells (group4) is at the bottom (Fig. 4b). Because of the well-captured mean-variance relationship, voomByGroup delivered well-controlled FDR compared to those from voom and voomQWB, especially when comparing between group1 versus group2 in scenario 3, where slightly higher technical variation is present (Fig. 4c).
Immune responses in asymptomatic COVID-19 patientsWhile the simulations allowed us to assess the performance of gold standard methods and group variance methods based on known truth, these results have no biological interest. Moreover, no matter how carefully thought-out and well-designed our simulations are, these data will inevitably miss some features from experimental data. Thus, we also examined the performance of methods on human scRNA-seq data.
Zhao et al. [32] investigated PBMCs from COVID-19 patients of varying severity alongside healthy controls (HCs), with a focus on the comparison between asymptomatic individuals and HCs. The study found that interferon-gamma played an important role in differentiating asymptomatic individuals and HCs, such that it was more highly expressed in natural killer (NK) cells of asymptomatic individuals [32]. In their data, the expression of IFNG was observed to be upregulated in asymptomatic individuals; however, the difference was not statistically significant when analyzed with edgeR QL. We reanalysed this dataset (PBMC1, see the “Methods” section) to see whether group variance methods could offer improved results.
We aggregated CD56dim CD16+ NK cells from each sample to create pseudo-bulk samples and then filtered out samples with fewer than 50 cells. A first glance at the data via MDS and group-specific mean-variance plots shows that HCs have a distinct mean-variance trend and less biological variation (Fig. 1c). By accounting for the relatively low variance in the HC group, we found that group variance methods outperformed gold-standard methods in terms of statistical power, such that they detected more DE genes for the comparison between HCs and asymptomatic individuals (Fig. 5a)—this is consistent with our simulation results when comparing groups with low variance (Fig. 3b). voomByGroup detected the most DE genes, followed by voomQWB: 880 and 719 genes respectively. The gold-standard methods, edgeR LRT, voom, and edgeR QL, detected 664, 453, and 403 DE genes respectively.
Fig. 5
Genes differentially expressed between NK cells from asymptomatic COVID-19 patients and healthy controls. a The number of genes DE in the comparison between CD56dim CD16+ NK cells from asymptomatic patients and healthy controls. Up means upregulated and down means downregulated in asymptomatic patients. Enriched GO terms related to interferon using DE genes detected with voomByGroup are plotted in panel (b). The x-axis displays the -log10 transformed p-values for the different Gene Ontology terms, and the color-scale also varies by p-value
To understand our results further, we looked at the consistency at which genes were detected as DE between methods (Additional file 1: Fig. S9a). We excluded edgeR LRT from our Venn diagram since the inclusion of all 5 methods greatly increased the complexity of the plot, and edgeR LRT was of less interest to us since we previously demonstrated that it performed poorly in the control of type I error. Although group variance methods detected almost double the number of DE genes as compared to voom and edgeR QL, most genes were detected by all methods (356 genes). Both of the voom-variants, voomQWB and voomByGroup, detected all of the genes that were also detected by voom. With the exception of 1 gene, voomByGroup also detected all of the genes that were detected by voomQWB. From voom to voomQWB then voomByGroup, the methods increase in their level of group-specific variance modeling. The overlap between these methods and the extra DE genes reflects the hierarchy in variance modeling for these methods and demonstrates the potential gain in statistical power when capturing group variance more accurately.
Next, we turned to Gene Ontology (GO) enrichment analysis to study the biological processes that play a role in COVID-19. We looked for any enrichment in GO terms for significantly upregulated genes in asymptomatic patients for each of the methods under examination. voomByGroup was the only method to detect the “interferon-gamma-mediated signaling pathway” as significant using a p-value cut-off of 0.01 (Fig. 5b). None of the other methods found any of these 5 genes as significant—they had much higher p-values as compared to that of voomByGroup (Additional file 1: Fig. S9b). To confirm the role of interferon-gamma in asymptomatic patients, we analyzed data from a separate study also involving CD56dim CD16+ NK cells in COVID-19 patients of varying severity [35]. The original study did not look into the role of interferon-gamma. Reanalyzing these data (PBMC2, see the “Methods” section), we found that in this second dataset the “interferon-gamma-mediated signaling pathway” was enriched using any of the DE methods under examination, and those group-specific variances were similar between all groups (Additional file 1: Fig. S10).
Taking the two COVID-19 datasets into consideration, we noticed a few things: (1) group variances can change between one dataset and another, even for studies on similar cell types and similar subjects—this perhaps has to do with how samples are processed (technical variation) and/or the “grouping” criterion plus the individual subjects involved (biological variation); (2) when variance trends are not too distinct from one another, all methods perform similarly, as observed in the second dataset (PBMC2); (3) when variance trends are distinct, group variance methods may benefit from a gain in statistical power, as observed in the first dataset (PBMC1); and (4) by modeling group-specific variances closely, voomByGroup was the only method that obtained statistically significant results for the biological process of interest in both datasets. These two datasets are used here to highlight how results of biological interest may be “missed” if heteroscedasticity is not carefully considered.
留言 (0)