
記住我
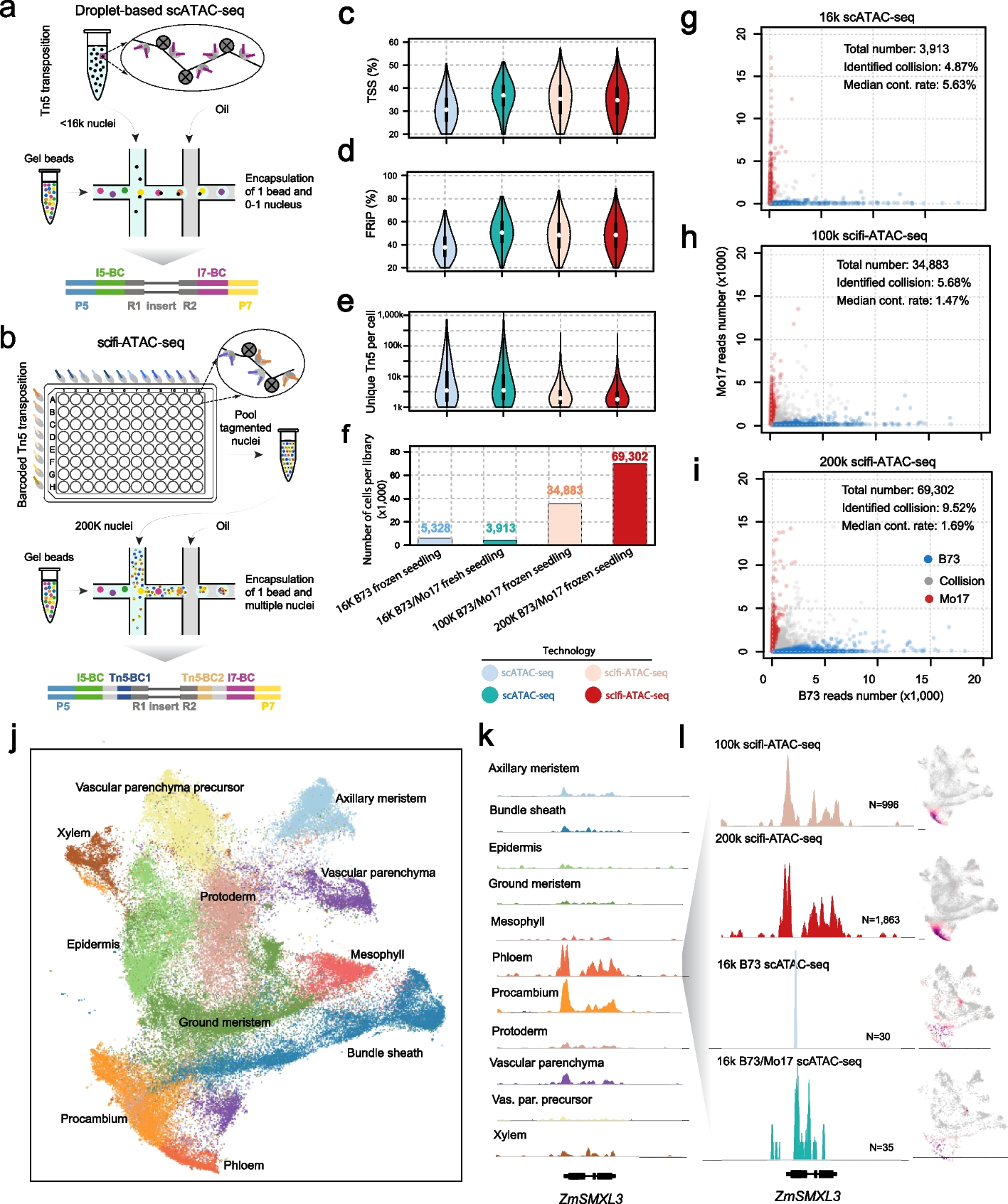
Plasma cfDNA was extracted from preoperative (Pre-OP) and postoperative (Post-OP) blood draws of 85 stage I-II CRC patients (Table 1) undergoing curative surgery. In addition, data from two stage III CRC patients were used in the model training. A biopsy from the resected tumor and paired peripheral blood cells was sequenced to generate a patient-specific mutational catalog. Post-OP samples were collected 2–4 weeks after surgical removal of the primary tumor (Fig. 2). Each cfDNA sample was sequenced using a custom hybrid-capture panel, designed to capture 41 exonic regions, spanning 15.413 bp, frequently mutated in CRC (Additional file 1: Section S1 and Additional file 1: Table S1). After collapsing UMI groups, the median of the average depths with corresponding interquartile range (IQR) of samples were for Pre-OP, 3307 (IQR: 3560); Post-OP, 7143 (IQR: 8844); buffycoat, 1850 (IQR: 1468); and tumor samples, 2132 (IQR: 2145); no samples had an average read depth below 100. All samples have been mapped and processed through the same pipeline (Additional file 1: Section S1).
Table 1 Clinical characteristicsFig. 2
The data collection setup for tumor-informed relapse detection in colon cancer patients. After the patient is diagnosed, a liquid biopsy is extracted prior to curative surgery (Pre-OP). A biopsy is taken from the resected tumor. Following surgery liquid biopsies (Post-OP) can be collected to monitor relapse. All collected samples are sequenced using next-generation sequencing
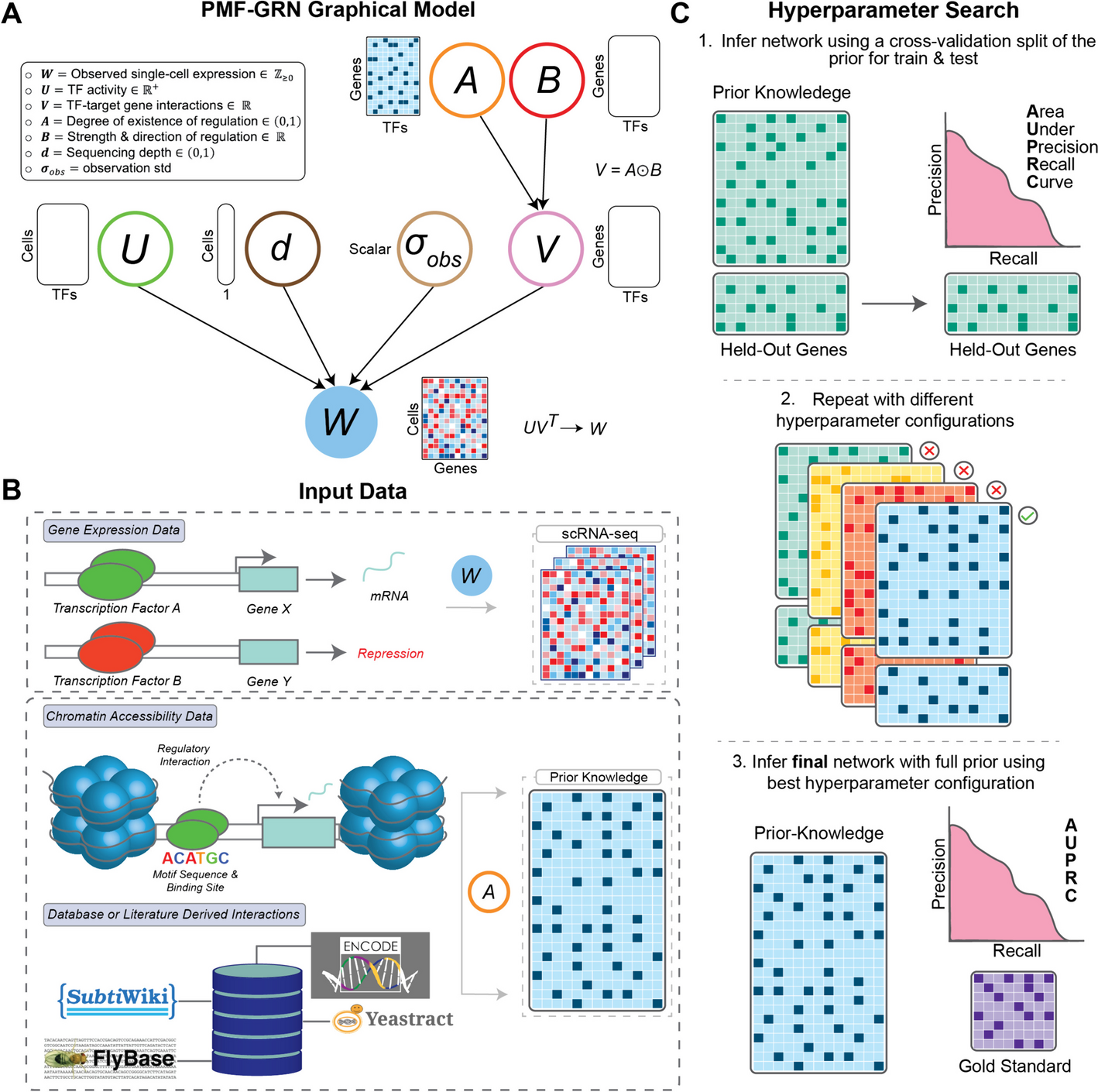
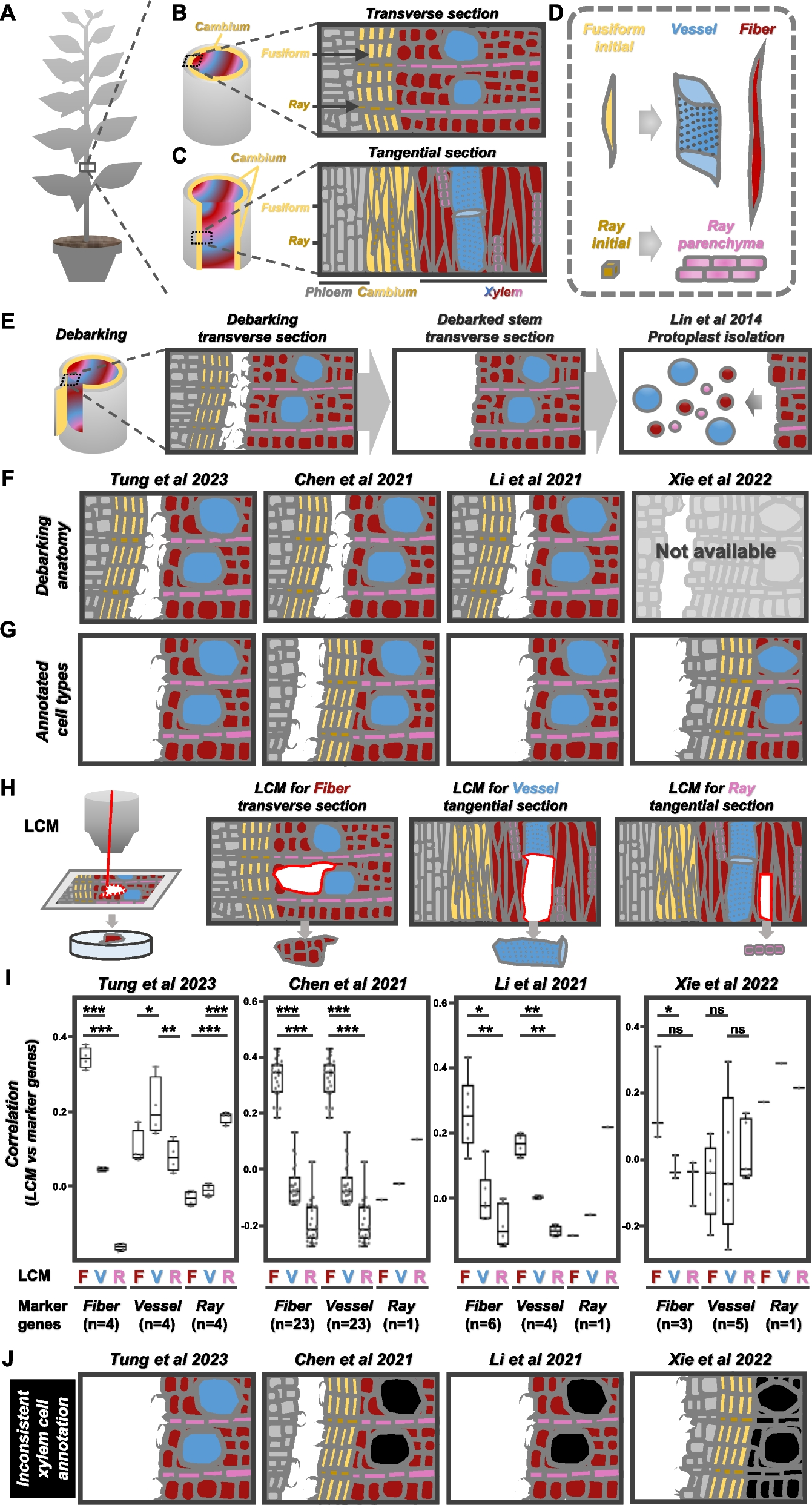
We first identified features that are known or expected to affect the error rate (Fig. 3a). In general, they can be split into two types: local sequence-context features and read-level features. The local sequence-context features capture the genomic sequence context, including the trinucleotide context, information about the sequence complexity (Shannon entropy of nucleotide, 1-mer, or dinucleotide, 2-mer, frequency), and GC contents in an 11 bp window around the position of interest (see the “Methods” section).
Fig. 3
a Examples of local sequence-context features and read-level features extracted from reads covering a position of interest in a read mapping. Centered at the position of interest, the trinucleotide context is extracted, and the surrounding 11-bp region is used for calculating regional features, including GC content and K-mer complexity. The read pairs contain a forward and reverse read that are enumerated as either the first or second of the pair according to the order of sequencing. Two read pairs are used for illustration of the read-centric features in the panels on the right. The UMI groups are shown to indicate the variation in the number of reads used for the consensus reads. The read position and fragment size are shown for the consensus reads. b–e Variation in observed error rate for selected features based on their observed distribution: b fragment size, c UMI group size, d read position relative to the start and end of reads and the variation between the first and second read in a pair. e Error type for each strand (forward and reverse). For each feature, the 95% confidence interval is indicated by the shaded areas or error bars. See Additional file 1: Section S3 for how the error rates and confidence intervals are calculated and similar plots of the remaining features
The read-level features capture the structural composition of the read, UMI characteristics, and sequencing information. The structural composition includes the strand a read aligns to (forward or reverse), the number of insertions and deletions in the read, and the total size of the underlying fragment. In the read pre-processing, UMIs were used to generate consensus reads with lowered error rates (Additional file 1: Section S2). For each consensus read, we extracted the UMI-group size, the number of reads disagreeing with the consensus at the position, and the overall number of mismatches outside the position of interest. As sequencing-related features, we included the base position in the read (read position), the length of the read sequence after overlap trimming, and whether the read is the first to be sequenced from the read-pair. The read quality (PHRED score) was not included, as it had the same high value for all positions in the UMI-collapsed consensus reads.
We evaluated the individual feature associated with the error rate by analyzing the total set of read alignment mismatches (n = 707,562) across all Post-OP samples (Fig. 3b–d), after excluding mutations and variants found in matching tumor and germline samples. The mismatches were compared to an equal number of randomly sampled matches, to estimate the error rate for each feature across its values (Additional file 1: Section S3).
Since fragment lengths of cfDNA are influenced by nucleosome binding patterns, the fragment length distribution has peaks at around 162 bp (mono-nucleosomal) and 340 bp (di-nucleosomal) [23]. The error rate tended to be minimized in fragments of these lengths (Fig. 3b). As expected, we observed a lower error rate in consensus reads formed by larger UMI groups [12] (Fig. 3c).
The error distribution for the read position showed an increased error rate in the beginning and end of the reads (Fig. 3d). We also observed a clear difference in error distribution along the read between the first and second read of the pair. The 12 different nucleotide alterations showed widely different error rates (Fig. 3e), which is expected as error-induced mismatches are not equally likely, and the rate further differed between the two strands. However, strand symmetric alterations were generally similar, apart from the mismatches C➔T/G➔A and C➔A/G➔T.
Overall, we saw variation in the error rate for all the presented features (the remaining are shown in Additional file 1: Section S3). Thus, for a given genomic position, different reads may have different error rates due to differences in read-level features. In the following, we present how this variation can be captured and used to potentially improve detection of ctDNA.
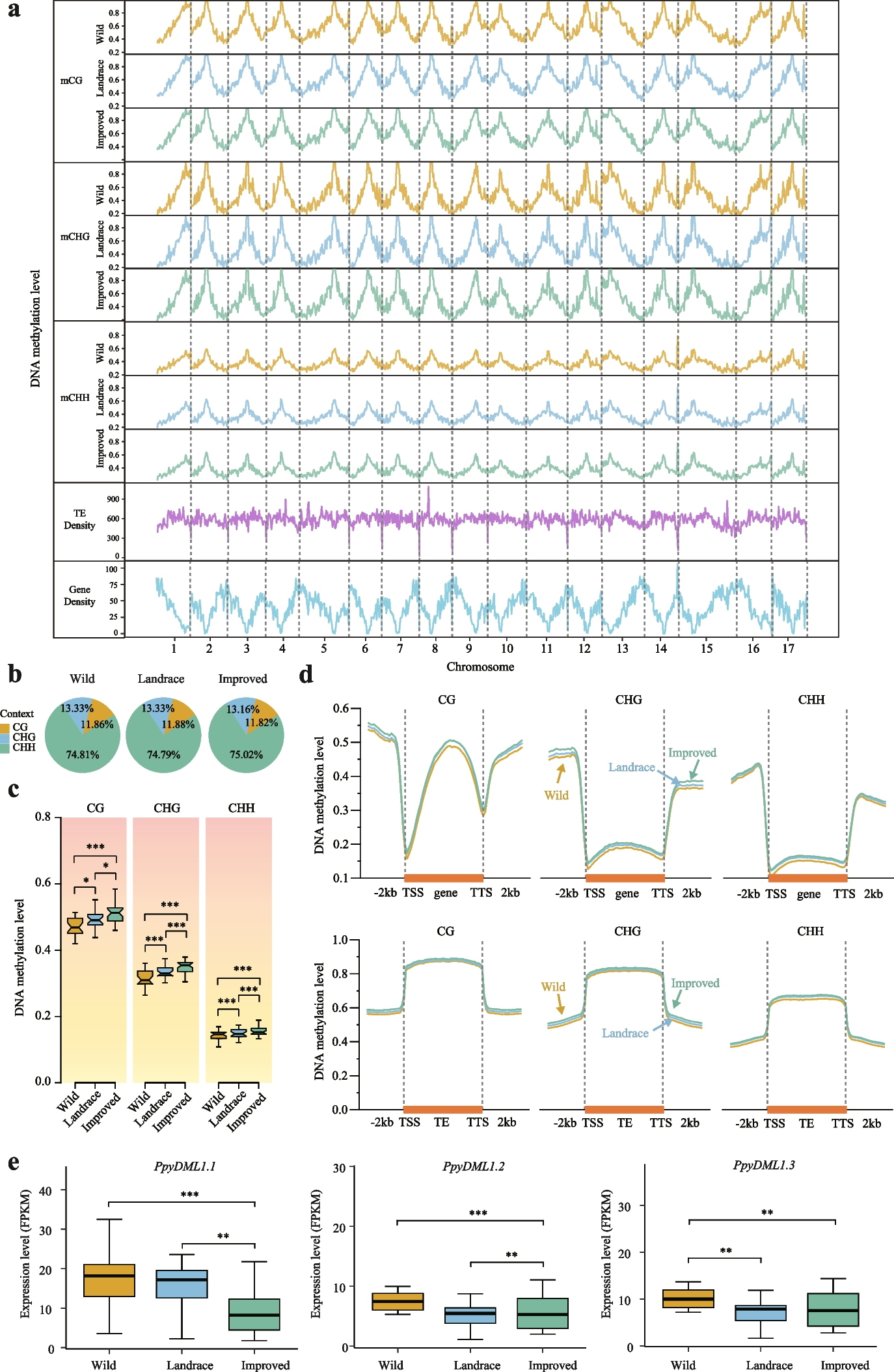
Neural network model and feature selectionTo predict the error rate at a given read position, we used a neural network model with the input features described above (see the “Methods” section). The predictive ability of individual features was evaluated using a “leave-one-covariate-out” (LOCO) scheme [24] (Additional file 1: Section S4). In short, we evaluated the performance of a full model containing all features (baseline) and then the relative performances of restricted models where each feature had been left out one by one. We used the latter to measure and rank the importance of each feature (Fig. 4a). When leaving out the trinucleotide context, the reference base was provided instead to assess only the importance of the two neighboring nucleotides.
Fig. 4
a Features are individually removed one-by-one from the full model containing all features to measure the decrease in validation error. The most important feature is then defined as the one that if removed decreases the validation error the most, and vice versa. The gray points show the mean decrease in validation error for each fold of a fivefold cross-validation. The average of these is used to rank the features by importance, indicated by the black points. b Based on the importance ranking, the features are cumulatively removed one-by-one from a full model. If the decrease in validation error compared to the full model is significant, the feature should not be removed from the model. A feature is only kept if removing it worsens the performance in all folds of the fivefold cross-validation. c Structure of the neural network model. The neural network uses three different types of input features: numeric, categorical, and embedded. The input features are processed differently in each group. The input features are then parsed through three hidden layers of decreasing width. The output contains 4 nodes representing the probability of observing each of the four based (A, T, C, G) at the given read position. d Illustration of 5 × 2-cross-validation procedure for the estimation of performance. The patients are first split into two approximately equally sized folds. The neural network model is trained on the Post-OP data of fold 1 and validated by testing the models on the Pre-OP samples of the other fold. This is then repeated by swapping the data in fold 1 and 2. The whole process is repeated 5 times
We found the most and second most informative feature for modeling the error rate to be respectively the read position and the strand (Fig. 4a). The third feature was the trinucleotide context, indicating that there is a difference in error rate for different contexts, as found by others [19]. The fourth feature was whether the read was the first in the read pair to be sequenced, indicating that systematic errors might be induced by the lab protocol. The fragment length, sequence length, and UMI group size also contribute significantly to the model. The remaining features showed little to no effect on the model performance. This showed that read-level features do contribute to accurate modeling of the error rate, and that they might be at least as important as features derived from the local sequence context.
An optimal subset of informative features was chosen using a stepwise procedure where features were excluded in order of importance (see the “Methods” section). The set of features chosen was the smallest model that did not perform significantly worse than the full model (Additional file 1: Section S4). The five least important features could be removed without any significant negative effect on the performance (Fig. 4b). Of the remaining eight features, seven were read-level features, namely the number of other errors in the read, the fragment length, the sequence length, the UMI group size, and if the read was first in pair, strand, and read position. The only remaining local sequence-context feature was the tri-nucleotide context.
The numerical and categorical variables are processed differently in the neural network prior to the hidden layers (Fig. 4c). The numerical features are batch normalized, the categorical features are one-hot encoded, and the tri-nucleotide context is embedded in three dimensions to handle the large number of possible contexts (see the “Methods” section).
To validate the utilization of the DREAMS error model, we applied it in calling tumor variants (DREAMS-vc) and cancer (DREAMS-cc) (see the “Methods” section). We assessed the performance using five repeats of twofold cross-validation (5 × 2 CV) (Fig. 4d). The model was trained on the Post-OP samples, and Pre-OP samples were used for method validation. The tumor variant positions were excluded from the training data. Potential signals from sub-clonal variants that are not detected in the tumor should be infrequent and present at low levels and therefore have minimal effect on the error model. By excluding variants found in the germline samples, we expect to reduce the potential signal of clonal hematopoiesis of indetermined potential (CHIP). The split was done on patient level to ensure that a model is not trained and tested on data from the same patient. This analysis was repeated with five different randomized splits to control for split-induced variation.
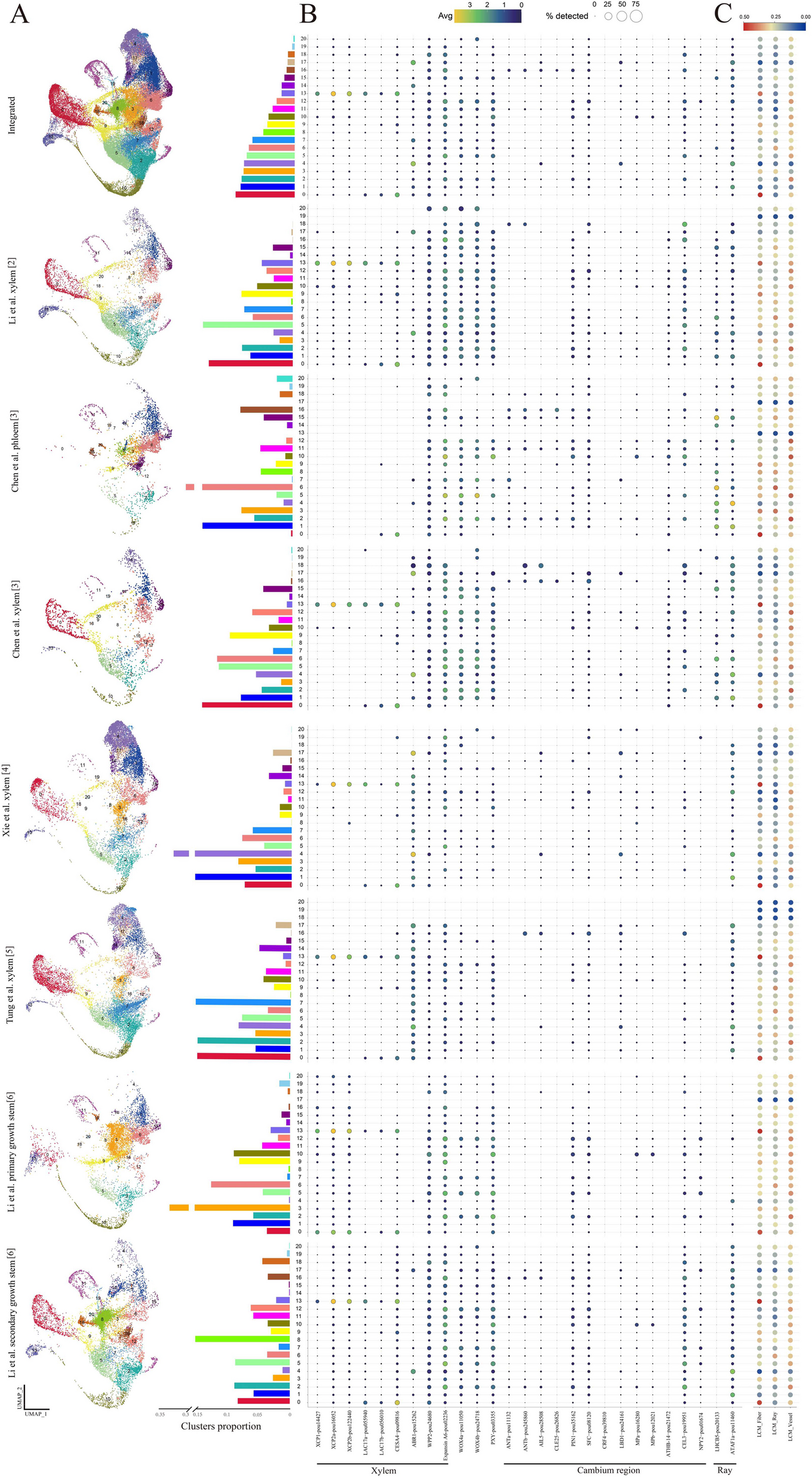
Predictive performance in clinical dataThe performance of calling tumor mutations in the plasma samples was assessed by looking at the area under the receiver operating characteristic curves (AUC). The performance of DREAMS-vc was compared to state-of-the-art algorithms Mutect2 and Shearwater. All positions with at least one observed mismatch were scored with each method and included in the performance calculations (Fig. 5a). Positions without signal was called negative by any method, making them redundant for performance comparisons. DREAMS was also compared to iDES [12] and Strelka2 [16]; however, these methods are not directly comparable and are therefore presented in Additional file 1: Section S5. In addition, we evaluated how the performance of DREAMS depended on the size of the training set and found it increased with the number of training samples, with the largest gain seen when going from one to two samples (Additional file 1: Section S6).
Fig. 5
a Performance of variant calling using DREAMS-vc compared to state-of-the-art tools Shearwater and Mutect2. The AUC is estimated based on the different negative sets: The cross-patient calls, 500 random validation alterations, and these sets combined (All). The AUC is also estimated for the full group of patients (All), and the patients with stage I and stage II CRC, individually (ns: p ≥ 0.05, *: p < 0.05, **: p < 0.01, ***: p < 0.001, ****: p < 0.0001). b Performance of variant calling using DREAMS-vc, Shearwater, and Mutect2 with various trimmings of the reads (start:end). The performance is compared pair-wise to the 0:0 model, with worse (red) and improved (green) performances indicated
Using DREAMS-vc, we aimed to call the tumor mutations of each patient from their respective mutation catalog. As negative controls, we attempted to call cross-patient mutations, by searching for the mutations found in other patients. Additionally, within the sequencing panel, a set of 500 randomly selected positions each associated with a random alteration, was used as negative controls, and referred to as validation alterations.
Evaluating across the combined negative set of both cross-patient mutations and validation alterations and cancer stages, DREAMS-vc performs significantly better than both Shearwater and Mutect2 (Fig. 5a). Additionally, the performance was assessed separately for stage I and stage II CRC patients. This showed superior performance of DREAMS-vc especially for the stage II CRC patients (Fig. 5a). As expected, all models perform better on later-stage patient samples as these are expected to have a higher mutational signal in the cfDNA due to a higher tumor burden. The methods perform more similarly on stage I patients; however, DREAMS-vc has significantly better performance.
Performance evaluations for each of the separate negative sets showed that DREAMS performs better than Mutect2 with the cross-patient negative set and better than Shearwater with the validation set as the negative set. The variation in performance of DREAMS-vc across splits and folds is lower than for Mutect2 and Shearwater, which indicates that its variant calling is more stable across patients and mutation types.
The explorative feature analysis indicated an increased error rate at the beginning and end of reads (Fig. 3d). DREAMS takes the read position into account and can thereby compensate for the increased error rate at these positions. Other methods such as Mutect2 and Shearwater are not aware of read ends and the performance of these can potentially improve by trimming these. To investigate the effect of trimming, we evaluated the performance of each method when trimming 0, 2, 6, or 12 of the bases in the beginning of reads or 0, 1, or 2 of the bases in the end of reads (Fig. 5b). We found that Shearwater can improve performance by trimming 2 bases from the beginning of reads and Mutect2 can improve by trimming the last base of each read. For DREAMS, the performance is only decreased when trimming, especially in the beginning of reads.
By setting the false-positive rate at 5% for each method across the validation alterations with any support, we get comparable thresholds for the three confidence measures: p-values, Bayes factor, and TLOD for DREAMS-vc, Shearwater, and Mutect2, respectively. This allows for a comparison of the sensitivity of the models at a pre-determined specificity of 95%. The methods could then be assessed across an alteration catalog of 191 true-positive mutations from the mutation catalog and 12,900 cross-patient negative calls based on the mutation catalog of the other patients. Out of the alteration catalog, 88 true mutations and 1330 cross-patient negative calls had a signal for the alteration.
Using this threshold, DREAMS-vc called 84.1% of the tumor mutations with signal, while Shearwater and Mutect2 called 70.5% and 67.0%, respectively (Table 2). F1 and G-mean scores were calculated to assess the performance of the models by using the cross-patient mutations as negative controls. G-mean is the geometric mean of sensitivity and specificity, and F1 is the harmonic mean of precision and sensitivity. For G-mean, DREAMS-vc performed better than Shearwater and Mutect2; however, the F1 score of Shearwater was very similar to DREAMS-vc, due to lower false-positive rate of shearwater (Table 2). Considering all mutations observed in the tumors, including those without signal in plasma, we found that about 38.7% could be recalled in Pre-OP liquid biopsy samples. DREAMS is generally more sensitive than Mutect2 and Shearwater, especially for stage II cancer mutations. Higher stage cancer is expected to have a larger signal for cancer mutations. Mutations called by DREAMS-vc but missed by Shearwater generally have low allele frequencies (Additional file 1: Section S7).
Table 2 Mutation calling performance (true-positive mutations and cross-patient negative calls)By setting the threshold based on a 5% false-positive rate in the cross-patient mutation set, the validation mutation set can be used as negative controls. The true positives are still the same 191 mutations of which 88 has a signal for the alteration. The negatives are the 500 validation positions multiplied with the 87 tested samples, giving a total of 43,500 possible alterations of which 1920 had a signal. With this set, we obtained an 86.4% true positive rate, compared to 76.1% for Shearwater and 67.0% for Mutect2 (Table 3). DREAMS-vc scored highest in both F1 and G-mean scores. Here, DREAMS-vc performed distinctly better than Shearwater and Mutect2.
Table 3 Mutation calling performance (true-positive mutations and validation alterations)A common measure used to predict the presence of ctDNA is the estimated tumor fraction in plasma. DREAMS-cc combines the mutational evidence across the mutation catalog, to estimate the tumor fraction with an accompanying p-value for the presence of cancer (see the “Methods” section). We aimed to detect cancer in the Pre-OP samples, since cancer is present and should, in theory, be detectable given enough ctDNA is present in the blood. As a negative control, we attempted to detect cancer in each Pre-OP sample (Tested Sample) with the mutation catalog from all other patients (Candidate patient) (Fig. 6a). In case of shared mutations between the mutation catalogs, these were eliminated to prevent false positives. As a benchmark, we constructed a cancer call score using the product of the individual Bayes factors across the mutation catalog from Shearwater, resulting in a similar tendency (Fig. 6b), and for Mutect2, we used the highest scoring variant, since we do not have a natural way of combining the scores (Fig. 6c). The performance of calling cancer can be assessed by treating the cross-patient mutation catalogs as expected negatives and calculate an AUC score (Fig. 6d). Performance was compared using the 5 × 2 cross-validation setup as above (Fig. 4d). The AUC was similar between DREAMS-cc and Shearwater with respect to calling cancer; however, DREAMS-cc showed an increased performance. Mutect2 does not seem to be suited for cancer calling in this way. As for variant calling, we only included the samples with mutational signal to showcase and compare the performance of the different methods in discriminating tumor from error signal.
Fig. 6
Prediction of cancer using a DREAMS-cc, b Shearwater, and c Mutect2. For each patient’s LB-sample (y-axis), the mutation catalog (x-axis) for every candidate patient is used for calling cancer. The patients are stratified into patients with stage I and stage II CRC. The diagonal is showing the result of using a patient’s own mutation catalog for cancer calling and constitutes the expected positives. The off-diagonal is the cross-patient results, for which the mutation catalog is filtered with the patient’s tumor and germline variants prior to cancer calling, and thus these are expected to be negative. The color scheme is chosen based on the matched quantiles of the rank from the p-value, combined Bayes factors and max TLOD from a DREAMS-cc, b Shearwater, and c Mutect2, respectively. The cancer predictions show the results from one split in the 5 × 2 CV. d AUC performance of DREAMS-cc, shearwater, and Mutect2 with respect to calling cancer using the 5 × 2 CV
For the patients with stage I and II CRC, we found tumor-supporting reads in 47.5% (19/40) and 73% (33/45) of the Pre-OP samples, respectively. We called cancer in 36.6% of the stage I CRC patients, corresponding to 78.9% (15/19) of the patients with a mutational signal. We called cancer in 72.1% of the stage II CRC patients, corresponding to 93.9% (31/33) of the patients with signal. These results were obtained while still limiting the false-positive rate to 5% in cross-patient cancer calls with a non-zero mutational signal.
Detailed analysis of the false-positive cancer calls reveals that most are due to a specific KRAS G12V variant: chr12:25,245,350 C > A. This variant is common in colon cancer, and it is therefore not surprising to find in the patients [25]. However, the mutation was not found in the patient’s corresponding tumor or buffycoat samples. A possible explanation for this is that the mutation is not detected in the tumor biopsy due to sub-clonality [26] or that there is an underlying germline signal that was not caught in the buffycoat.
留言 (0)