
記住我
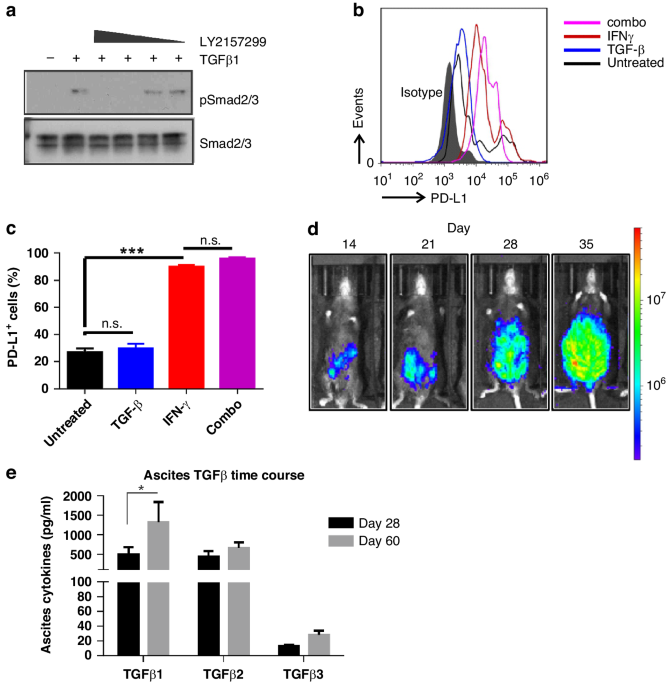
A total of 107 patients were enrolled in the study, including 18 patients across 13 dose-escalation cohorts and 89 patients in expansion cohorts; though eligible for enrolment, no patients with anal cancer were enrolled in the study. Five patients were concurrently enrolled in both dose-escalation and expansion cohorts (Fig. 1).
Fig. 1: SRA737 monotherapy study enrolment by dose level.
Patient numbers and tumour types enrolled in different cohorts in dose escalation and expansion phases of the study.
In the Dose Escalation phase the patients described as having other disease included pancreatic cancer (1), mesothelioma (2), esophageal carcinoma (1) and cholangiocarcinoma (1). In the Expansion Phase the patients described as having other disease included fallopian tube carcinoma (1) and oropharyngeal carcinoma (1).
The median age of patients in this study was 64 years (range 38–86 years) and the most common tumour types were HGSOC (n = 37), CRC (n = 32), and mCRPC (n = 16). Within the study, 98% (105/107), 41% (44/107) and 36% (38/107) had previously received chemotherapy, immunotherapy or radiotherapy respectively. In addition,100% (16/16) and 24% (9/37) of patients with prostate cancer and ovarian cancer had previously received hormonal therapy and PARP inhibitors respectively. More than 50% of patients had received four or more lines of previous treatment (Supplementary Table 2).
Patients enrolled in tumour-type expansion cohorts were required to have genetic alterations hypothesized to confer sensitivity to Chk1 inhibition. Of the pre-specified alterations required for eligibility, the most frequently detected were tumour suppressors TP53 (56% of patients), PTEN (15% of patients) and CDKN2A/B (8% of patients); DNA damage response genes BRCA2 (8% of patients) and ATM (7% of patients); and oncogenic drivers KRAS (23% of patients), FBXW7 (9% of patients), and MYC (8% of patients).
Safety profileDose escalation and tolerabilityThe study enrolled a total of 18 patients in the dose-escalation phase and followed a single-patient dose escalation strategy at SRA737 dose levels of 20, 40, 80, 160, 300, 600 and 1000 mg QD. No DLTs were seen in the 1000 mg QD dose-escalation cohort (or any lower-dose cohorts), and thus the expansion cohorts were opened. Safety data were reviewed when a total of 16 patients had been enrolled at the 1000 mg QD dose level (dose-escalation or expansion cohort); of the 10 evaluable patients, 3 patients required dose reduction to 600 mg due to GI intolerability and 7 patients tolerated the 1000 mg dose but required the support of anti-emetic medication and/or night-time dosing. A cohort evaluating the 1000 mg daily dose split into two doses (500 mg twice daily [BID]) then enrolled six patients, four of whom were evaluable for DLT. One patient in the 500 mg BID dose group had a DLT of Grade 4 thrombocytopenia, and review of all patients showed no clear differentiation in overall tolerability of 500 mg BID vs. 1000 mg QD dosing, thus there was no further enrolment in BID dosing. Three patients were enrolled into the 1300 mg QD cohort; 2 of these patients had dose-limiting GI intolerability (Grade 1–2 nausea, vomiting, and/or diarrhoea), and 1 patient had dose-limiting Grade 3 neutropenia. Therefore, the dose of 1000 mg QD was considered the MTD. Expansion cohorts were initially enrolled at 600 mg QD (5 patients), however, the expansion phase was predominantly enrolled at the 1000 mg QD and 800 mg QD dose levels (Fig. 1).
Overall safety profileThe most common treatment-emergent adverse events (TEAEs) attributed to SRA737 irrespective of SRA737 dose level were diarrhoea (63%), nausea (60%), vomiting (46%), fatigue (38%), decreased appetite (18%), neutropenia (16%) and anaemia (15%) (Table 1). Among the 49 patients treated at SRA737 doses >800 mg QD, the most frequent Grade 3 or higher TEAEs (aside from disease progression) related to SRA737 were neutropenia (10%), rash/rash maculopapular (6% combined), and fatigue (4%) (Table 2).
Table 1 SRA737-related treatment-emergent adverse events of any grade reported by ≥10% of overall patient group.Table 2 SRA737-related treatment-emergent adverse events of Grade 3 or higher reported by 2 or more patients overall.Cardiac safety parameters were monitored closely in this study since cardiac side effects have been seen previously with Chk1 inhibitors. The studied doses had no clinically relevant effects on heart rate, PR interval, or QRS duration and there were no individual subjects with treatment-emergent QTcF values >500 ms or a change from baseline in QTcF (ΔQTcF) of >60 ms on central ECG monitoring. Four cardiac serious adverse events were evaluated to be possibly related to SRA737. At 800 mg SRA737, 1 case of asymptomatic myocardial infarction was diagnosed by the detection of elevated troponins on routine monitoring concurrent with ECG changes and mild systolic dysfunction and apical akinesis on echocardiogram, and 1 case of acute coronary syndrome with complaints of chest pain associated with elevated troponin but no ischemic changes noted on ECG occurred. At 1000 mg SRA737, 1 case of cardiomyopathy manifested by chest pain and shortness of breath with echocardiographic findings of left ventricular systolic dysfunction and asymptomatic myocardial infarction, and 1 case of asymptomatic QT prolongation with no cardiac signs or symptoms reported including during a period of in-hospital observation occurred. In all cases, administration of the drug was permanently discontinued. Based on an analysis of the relationship of SRA737 plasma concentration and QT interval corrected for heart rate using Fridericia’s formula (QTcF) measured in 79 patients, the QTcF least squares (LS) mean change from baseline (ΔQTcF) was similar across all dose groups and indicated that SRA737 has an effect on QTcF. The largest LS mean ΔQTcF at the recommended Phase 2 dose level of 800 mg QD was observed at 2 h post dose on Cycle 1 Day 22, corresponding to the time of maximum plasma concentration (Tmax) of SRA737, and reached 16.8 ms (90% confidence interval: 11.53 to 22.13). As noted above, no clinically significant treatment-emergent QTcF values and no clinically significant changes from Baseline were noted in a review of QTcF data from centrally read ECGs. Furthermore, no cases where ECG data demonstrated an increase in QTcF were associated with an arrhythmia or were associated with other symptoms. These clinical data reveal no indication that SRA737 is proarrhythmic at therapeutic doses.
A review of echocardiogram data obtained on 83 evaluable patients revealed that no additional patients (other than the cases mentioned above of clinically evident cardiomyopathy and myocardial infarction) had a > 10% absolute drop from baseline resulting in abnormal left ventricular ejection fraction.
Adverse events (AE) leading to treatment discontinuation were reported by a total of 34 patients (31.8%), 16 of whom (15.0%) discontinued due to SRA737-related TEAEs. Fatal adverse events AE were reported for ten patients; none were attributed to SRA737.
Pharmacokinetic profileThe PK profile of SRA737 after repeated doses of 600, 800 and 1000 mg QD is presented in Table 3. The Tmax of SRA737 generally occurred at between 2 and 4 h but ranged between 1 and 6 h. Estimated elimination half-life (t½) values ranged from 6.23 to 10.4 h, individual apparent total clearance of the drug from plasma after oral administration (CL/F) values ranged from 18.4 L/h to 119 L/h, and individual apparent volume of distribution (Vd/F) values ranged from 215 to 1590 L. Systemic exposure, as estimated by maximum plasma concentration (Cmax) and area under the plasma concentration-time curve (AUC), tended to be comparable between 600 mg and 1000 mg dose levels.
Table 3 Pharmacokinetic parameters (mean ± standard deviation) for plasma SRA737 following repeated doses.Upon repeated dosing at 800 mg and 1000 mg QD, systemic exposure generally remained similar regardless of specific sampling day (Cycle 1 Day 22 [C1D22], C1D8, C2D8, C2D15, or C2D22) compared to the first single dosing at Day −7 to Day −4, with individual accumulation ratio (RAUC) values ranging from 0.611 to 2.11.
The mean minimum SRA737 plasma concentration (Cmin) at the recommended Phase 2 dose of 800 mg QD was 312 ng/mL, which exceeded the minimally effective concentration of 100 nM (37.9 ng/mL) previously shown to inhibit Chk1 in preclinical experiments. All patients at or above the dose level of 300 mg QD achieved this concentration for at least 24 h following a single SRA737 dose, and up to 48 h for the majority of patients (Fig. 2).
Fig. 2: Single dose pharmacokinetic profile of SRA737.
Mean plasma concentrations of SRA737 across different dose levels over a 48 hour sampling window. Note: Error bars indicate ±1 standard deviation.
Determination of recommended Phase 2 doseThe majority of patients in this study initiated SRA737 at either the MTD of 1000 mg total daily dose (44/107 patients) or a dose of 800 mg QD (43/107 patients). The choice of 800 mg QD as the recommended Phase 2 dose for SRA737 monotherapy was based on the clinical observation that GI tolerability appeared to be improved at 800 mg QD versus the MTD of 1000 mg QD. Looking specifically at the frequency of AEs at 800 mg and 1000 mg SRA737, Grade 1–2 TEAEs of diarrhoea, nausea, and vomiting occurred in 68%, 78%, and 50% of patients at the 800 mg SRA737 dose level respectively, whilst these occurred in 72%, 65%, and 63% of patients respectively at the 1000 mg SRA737 dose level. Events of GI toxicity above Grade 2 were observed in one patient at the 800 mg dose level who experienced Grade 3 diarrhoea, whereas at the 1000 mg dose level 1 patient experienced Grade 3 diarrhoea and another single patient experienced both Grade 3 nausea and vomiting.
Exposure at both 800 mg QD and 1000 mg QD exceeded the minimum effective concentration extrapolated from preclinical models.
Tumour responseEfficacy analysis was performed on expansion cohort patients with predefined genetic mutations who received at least 75% of the planned dose of SRA737 in Cycle 1; there were no complete or partial RECIST responses (Fig. 3). The disease control rate, defined as patients who received at least four cycles of therapy with at least stable disease as their best response, was 8/25 (33.3%) in CRC, 11/26 (42.3%) in HGSOC, 3/8 (37.5%) in NSCLC, 8/13 (61.5%) in mCRPC, and 3/4 (75.0%) in HNSCC.
Fig. 3: Clinical efficacy of SRA737.
A waterfall plot of change in size of tumours represented as percentage at baseline in patients treated on the expansion cohorts. The genomic profile of individual patients are shown on the chart below the waterfall plot.
The lack of partial or complete RECIST responses precluded evaluation of associations between tumour response and genetic alterations, however, genetic aberrations of patients in expansion cohorts are displayed in Fig. 3.
留言 (0)