Human heme oxygenase 1 (hHO-1) is an enzyme that is overexpressed in response to heat shock, oxidative stress, presence of free heme, heavy metals, and other stimuli and pathways [1]. It is typically recognized for its crucial role in cell physiology with respect to its role in free heme degradation [[2], [3], [4], [5]]. The enzyme degrades heme to biliverdin with release of iron and carbon monoxide, and the biliverdin is subsequently converted to bilirubin by biliverdin reductase [2,[6], [7], [8], [9]]. The products of the degradation reaction, biliverdin and bilirubin, are reported to have antioxidant properties against hydrogen peroxide, peroxyl and hydroxyl radicals as well as anti-inflammatory properties [2,10]. The final product, bilirubin, decreases oxidative injury caused by hydrogen peroxide [11], can protect vitamin A and linoleic acid from degradation [12] and acts against reactive oxygen species which have been linked to cancer, aging, and inflammation progression [12]. While it is well established that human heme oxygenase has a role in fighting oxidative stresses and protecting the cells from oxidative damages, [1,2,12] its cytoprotective nature has been utilized by cancer cells for self-protection against chemotherapy, giving tumors resistance to standard treatments [1]. Hence, the enzyme has been linked to cancer progression and therapeutic resistance [1,13,14].
Accordingly, hHO-1 presents a promising target for cancer treatments. Various approaches, mainly genetic or pharmacological, can be employed for inhibition of the enzyme [15]. Genetic inhibition entails use of CRISPR/CAS9 or RNAi to specifically target the hHO-1 gene. Genetic techniques have the advantage of targeting the gene or mRNA directly, albeit they could increase the chances of off-target effects [15]. The alternative pharmacological approaches entail the use of metalloporphyrins like zinc protoporphyrin IX or azole-based drug inhibitors. Although metalloporphyrins have strong activity for inhibition, their potentially lower selectivity poses some risk factors [15]. On the other hand, azole-based compounds prove to be strong and more selective inhibitors; however, their drug metabolism and pharmacokinetics profiles (DMPK) need to be further explored [15].
Comprehensive reviews on the development of azole-based inhibitors for hHO-1 inhibition have previously been presented [15]. Development of the inhibitors initially started from a lead structure of azalanstat, a non-porphyrin inhibitor of 14α-demethylase cytochrome P450 (CYP51) [9]. These types of inhibitors have the unique advantage of acting through non-competitive inhibition, unlike metalloporphyrins which are competitive with respect to the heme. However, they present potential side reactivity and off-target selectivity issues, particularly with several physiologically relevant heme-containing proteins like cytochromes P450s, globins and other heme enzymes [1]. The development of these inhibitors finally led to azole drugs with a unique pharmacophore previously described by Rahman et al. [1] which is based on the “initial” azalanstat structure and the previously defined regions of interest comprised of a central region containing a dioxolane group, a bulky northeastern group, an imidazole that binds heme in the eastern region, and a western chlorophenyl group [1].
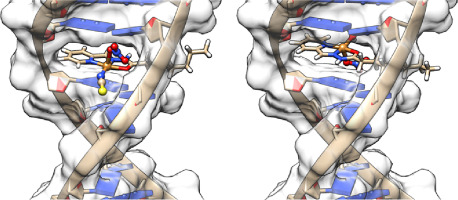
Since enzymatic inhibition can have profound physiological consequences, structure-function correlation studies of the protein in the presence of an inhibitor are of great importance. Resonance Raman (rR) spectroscopy is one of the most powerful tools for studies of the active site structure of heme proteins. rR spectroscopy has previously been successfully utilized to investigate the interactions between inhibitors with cytochromes P450; e.g., studies of CYP3A4 in the presence of N-carbene inhibitor showed its effects on heme planarity, ruffling, and interplay with the enzymes active site residues [16,17] CYP102 in the presence of a nitrogen-coordinating inhibitor revealed alteration of heme modes [18]. Furthermore, rR studies of CYP2B4 in the presence of acetylenic inhibitor 4-(tertbutyl) phenylacetylene (BPA) and BPA-modified CYP2B4 showed remarkable potential of rR spectroscopy to effectively monitor structural changes associated with inhibition, not only in ferric state but also in the ferrous CO-ligated form [ 18,19]. In this work we provide detailed spectroscopic insight into the interactions of three azole based inhibitors with human heme oxygenase. All inhibitors studied here, that is, compounds E, X and OB-24 (Fig. 1), contain an imidazole moiety that was previously shown to enable inhibition of hHO-1 [1]. The previous work with the OB-24 showed improved immunotherapy efficiency in treating tumor cells [13]. X also exhibited inhibitory function and compound E was chosen based on the predicted function owing to its structural similarity to OB-24 [1]. The equilibrium binding studies and rR measurement of hHO-1 in its ferric, ferrous and ligated states are complemented by molecular docking calculations.




留言 (0)