
記住我










TP0473292 is an adamantane carboxylic acid (ACA) ester prodrug for enhancing the oral bioavailability of the hydrophilic glutamate analog TP0178894, a novel metabotropic glutamate 2 and 3 receptor antagonist, and being developed as an antidepressant. TP0473292 showed high membrane permeability and rapid hydrolysis to TP0178894 in rat, monkey, and human liver S9 fractions, with a conversion rate of such that complete conversion by first-pass metabolism was expected. TP0473292 was also hydrolyzed in the intestinal, renal, and lung S9 fractions, coinciding with the result that TP0473292 was activated by carboxylesterase (CES) 1 and more efficiently by CES2. Despite the rapid hydrolysis of TP0473292 in the intestinal S9 fraction, TP0473292 achieved good oral bioavailability of poorly permeable TP0178894 (approximately 60%) in rats and monkeys, with no TP0473292 detected in the plasma, revealing that rapid hydrolysis in the intestine is not necessarily a disadvantage. We also confirmed the penetration of TP0178894 into the cerebrospinal fluid and its unmetabolized excretion in urine. The ester promoiety, ACA, was metabolized to chemically stable acyl glucuronide and excreted in urine in rats and monkeys, suggesting a low risk of idiosyncratic drug toxicity. TP0473292 and its metabolites did not show a drug-drug interaction potential via cytochrome P450 in humans. These results suggested that TP0473292 functions as an ideal oral prodrug in humans; this was later confirmed to be true in phase 1 clinical trials. Furthermore, ACA was firstly confirmed to be a useful promoiety for hydrophilic drugs to enhance their oral bioavailability.
SIGNIFICANCE STATEMENT Hydrolysis in the intestine reportedly has negative effects on the oral bioavailability of hydrophilic active metabolites of ester prodrugs. This study reports the preclinical pharmacokinetics of a hydrophilic metabotropic glutamate 2/3 receptor antagonist, TP0178894, and its ester prodrug TP0473292, which was found to act as an oral prodrug despite being activated predominantly in the intestine. Furthermore, this study firstly reports that adamantane carboxylic acid is useful as the ester promoiety of a prodrug for increasing lipophilicity and oral bioavailability.
IntroductionAbnormalities of glutamatergic transmission have been implicated in the pathophysiology of depression, and the group II metabotropic glutamate (mGlu) 2 and 3 receptors have potential as targets for the treatment (Chaki, 2017). The mGlu2/3 receptor antagonists reportedly have rapid and sustained antidepressant-like effects in rodent models, similar to (R,S)-ketamine (Chaki, 2017; Witkin, 2020), whose stereoisomer esketamine is used as an adjunct therapy for treatment-resistant depression, with the similar neural mechanisms to (R,S)-ketamine (Chaki and Fukumoto, 2019) but not showing (R,S)-ketamine associated side-effects (Witkin, 2020). These findings suggest that mGlu2/3 receptor antagonists hold promise as future antidepressants.
We previously discovered potent and selective mGlu2/3 receptor antagonists MGS0039 (BCI-632; Fig. 1A) and (1R,2R,3R,5R,6R)-2-amino-6-fluoro-3-[(4-fluorophenyl)methoxy]bicyclo[3.1.0]hexane-2,6-dicarboxylic acid (TP0178894) (Fig. 1B) (Nakazato et al., 2004). These were hydrophilic glutamate analogs, which possibly led to a low membrane permeability and, thus, a low oral bioavailability, making it difficult to develop them directly as oral antidepressants. In fact, MGS0039 showed a low oral bioavailability in rats (10.9%) and monkeys (12.6%) (Nakamura et al., 2006). Therefore, we adopted a prodrug strategy to increase its lipophilicity and synthesized MGS0210 (BCI-838; Fig. 1A), an n-heptyl alkyl ester prodrug of MGS0039. MGS0210 showed a relatively high conversion rate to MGS0039 in monkey and human liver S9 fractions among several ester prodrugs that were evaluated and an improved oral bioavailability (38.6%) as MGS0039 in monkeys (Nakamura et al., 2006). However, although MGS0210 was certainly absorbed, it mainly circulated in the plasma as the unchanged form, with approximately 10-fold higher exposure than MGS0039 in humans (Gadient et al., 2012), making it difficult to describe as an ideal prodrug in terms of pharmacokinetics (Beaumont et al., 2003). A possible reason for the limited exposure to MGS0039 was that the conversion rate of MGS0210 to MGS0039 was not fast enough to achieve rapid and complete conversion. These results indicated that a successful oral prodrug would need to be converted much faster than MGS0210.
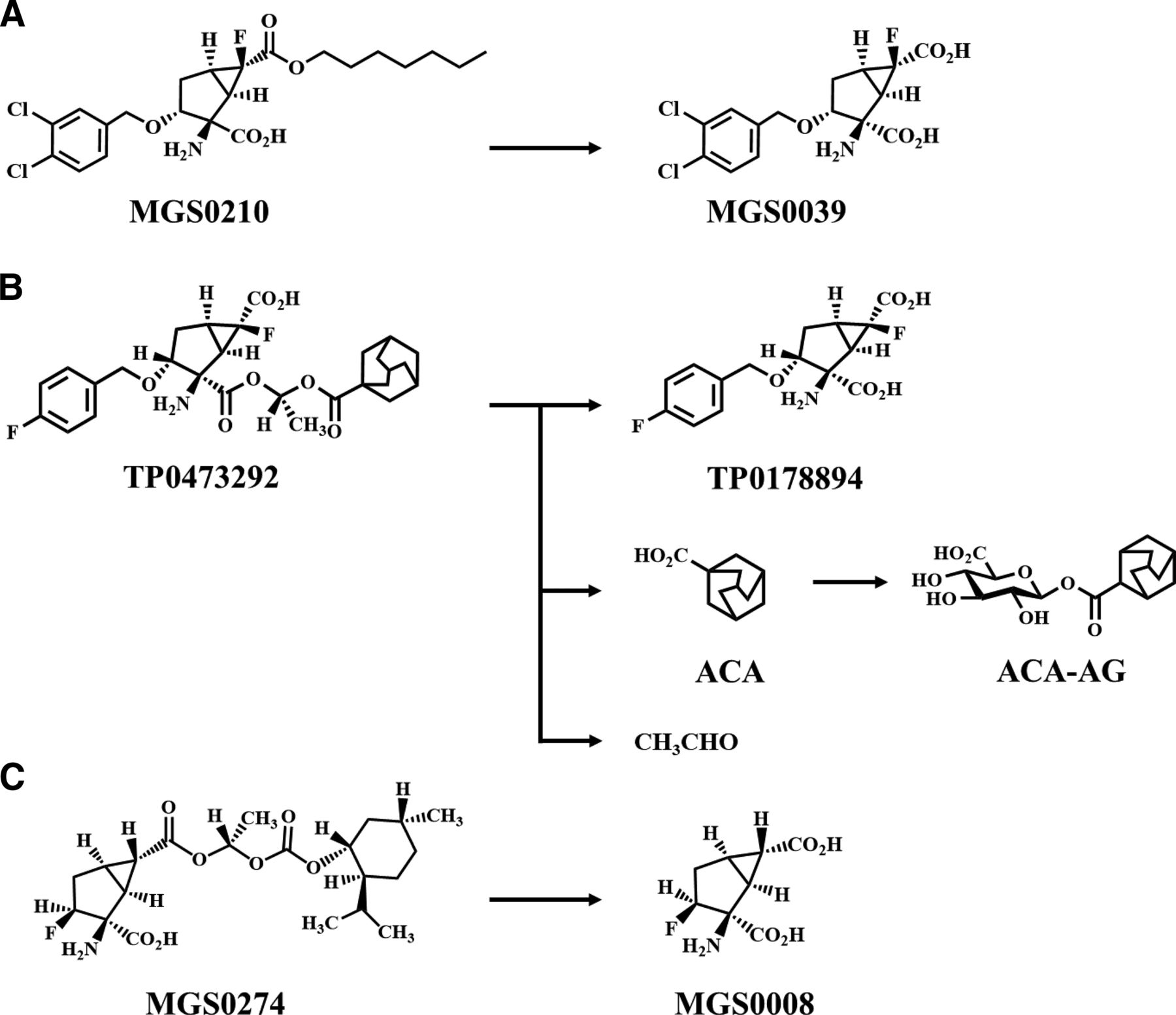 Fig. 1.
Fig. 1. Structures of ester prodrugs of glutamate analogs. (A) MGS0210: n-heptyl alkyl ester prodrug of an mGlu2/3 receptor antagonist MGS0039. (B) TP0473292: an adamantane carboxylic acid ester prodrug of an mGlu2/3 receptor antagonist TP0178894, shown with its proposed metabolic pathways in rat, monkey, and human cryopreserved hepatocytes. (C) MGS0274: an l-menthol ester prodrug of an mGlu2/3 receptor agonist MGS0008.
Subsequently, we confirmed this assumption by succeeding in the creation of MGS0274 besylate (Urabe et al., 2020; Fig. 1C), an l-menthol ester prodrug of an mGlu2/3 receptor agonist MGS0008 which was a glutamate analog and structurally similar to MGS0039 and TP0178894. MGS0274 exhibited a preferable lipophilicity and a higher conversion rate to its active metabolite than MGS0210 in monkey and human liver S9 fractions and achieved a high oral bioavailability (83.7%) as MGS0008 in monkeys with very low plasma exposure to MGS0274 (Kinoshita et al., 2019). In phase 1 clinical studies, MGS0274 was rapidly absorbed and extensively converted to MGS0008, whereas the plasma exposure to MGS0274 was low, approximately 3% of MGS0008 in a molar concentration, after oral administrations of MGS0274 besylate (Watanabe et al., 2020). Based on these results, we hypothesized that a prodrug with a high membrane permeability and faster conversion rate than MGS0274 would succeed as an ideal oral prodrug, even better than MGS0274.
Following this prodrug strategy, we synthesized a series of ester prodrugs of TP0178894 using various promoieties such as alkyl alcohols or l-menthol and selected (1R,2R,3R,5R,6R)-2-(carbonyl)-2-amino-6-fluoro-3-[(4-fluorophenyl)methoxy]bicyclo[3.1.0]hexane-6-carboxylic acid (TP0473292) (Fig. 1B) as the best. TP0473292 is a prodrug in which one of the carboxylic acids of TP0178894 is masked by adamantane-1-carboxylic acid (ACA) with two ester bonds to increase the lipophilicity. Adamantane is a promising moiety in drug design for providing lipophilicity to hydrophilic pharmacophores (Wanka et al., 2013). To apply an oral prodrug, adamantane should be conjugated with an active compound through a linker that is not chemically degradable, as in the case of a pH-sensitive linker in adamantane-conjugated doxorubicin prodrugs (González-Méndez et al., 2019), but is rapidly cleaved to release the active compound after absorption. Additionally, adamantane moiety should be cleared from the body without showing biologic activity, unlike amantadine prodrugs (Aboul-Fadl et al., 2011). Therefore, we used ACA as an inactive promoiety to conjugate adamantane with TP0178894 through two ester bonds, expecting not only increasing lipophilicity but also rapid enzymatic degradation. To our knowledge, there was no clinical prodrug that improves oral bioavailability of its active metabolite by adding adamantane through ester conjugation with ACA. Moreover, there was no report on the biologic fate of ACA as a liberated promoiety.
In this research, preclinical pharmacokinetic studies were conducted to evaluate whether TP0473292 could act as an ideal oral prodrug in humans. We also investigated the pharmacokinetic drug-drug interaction (DDI) potential of TP0473292 and its metabolites via cytochrome P450 (P450) in humans. Additionally, we firstly evaluated the usefulness of ACA as an ester promoiety for improving oral bioavailability of a hydrophilic compound.
Materials and MethodsMaterialsTP0473292, TP0178894, and 1-O-(tricyclo[3.3.1.13,7]decane-1-carbonyl)-β-D-glucopyranuronic acid (adamantane-1-carboxylic acid acyl glucuronide) (ACA-AG) were synthesized at Taisho Pharmaceutical Co., Ltd. (Saitama, Japan). TP0473292 was synthesized by the method disclosed in the patent application (Otake et al., 2017). TP0178894 was synthesized by the previously reported method (Nakazato et al., 2004). ACA-AG was synthesized in reference to the reported method (Monrad et al., 2014). ACA was purchased from Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan). TP0181164, the structural analog of TP0178894, and the stable isotope-labeled TP0473292, TP0178894, ACA, and ACA-AG used as internal standards were also synthesized at Taisho Pharmaceutical Co., Ltd. in similar methods as to TP0178894 and each unlabeled compound described above. [3H]TP0473292 was synthesized at Sekisui Medical Co., Ltd. (Tokyo, Japan). Benzil and bis(p-nitrophenyl) phosphate (BNPP) were purchased from Tokyo Chemical Industry Co., Ltd. Ethephon and EDTA dipotassium salt (EDTA-2K) dehydrate were purchased from FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan). Phenylmethanesulfonyl fluoride (PMSF) was purchased from Merck KGaA (Darmstadt, Germany). Male Sprague-Dawley (SD) rat plasma (anticoagulant: EDTA-2K) and serum were obtained from HAMRI CO., LTD. (Ibaraki, Japan) and The Jackson Laboratory Japan, Inc. (Kanagawa, Japan), respectively. Cynomolgus monkey plasma (anticoagulant: EDTA-2K) and serum were purchased from HAMRI CO., LTD. Human plasma (anticoagulant: EDTA-2K) and serum were obtained from healthy male and female volunteers after review and approval by the Ethics Committee of Taisho Pharmaceutical Co., Ltd. The rat, monkey, and human plasma and sera were stored frozen at −80°C until use. Rat and monkey cryopreserved hepatocytes and human liver microsomes were purchased from Sekisui Xenotech, LLC (Kansas City, KS), and human cryopreserved hepatocytes were obtained from BioIVT (Westbury, NY). Cryopreserved primary human hepatocytes from three donors (two males and one female) were purchased from Thermo Fisher Scientific (Waltham, MA). Rat, monkey, and human tissue S9 fractions of the intestine (PMSF free), liver, lung, and kidney were obtained from Sekisui XenoTech, LLC. Recombinant human carboxylesterase (CES) 1 and CES2 bactosomes were purchased from Cypex Ltd. (Dundee, UK). All other reagents were of analytical grade or of high-performance liquid chromatography grade and were obtained from commercial sources.
AnimalsMale SD rats (7 weeks old at the time of administration) were purchased from The Jackson Laboratory Japan, Inc. and were housed under controlled conditions with an approximate temperature (23°C ± 3°C), an approximate humidity (50% ± 20%), and lightning 12 hours/d at Taisho Pharmaceutical Co., Ltd. Male cynomolgus monkeys (8 years old at the time of administration; Simian Conservation Breeding & Research Center, Inc., Makati, Philippines) were housed under controlled conditions with an approximate temperature (24°C ± 3°C), an approximate humidity (50% ± 20%), and lightning 12 hours/d at Taisho Pharmaceutical Co., Ltd. Food was provided to rats ad libitum and monkeys once daily, except during fasting periods. Water was provided ad libitum. All the animal experimental procedures were approved by the Institutional Animal Care and Use Committee of Taisho Pharmaceutical Co., Ltd. and were performed in accordance with the 2006 Science Council of Japan Guidelines for Proper Conduct of Animal Experiments.
Parallel Artificial Membrane Permeability AssayThe membrane permeability of TP0473292 and TP0178894 at pH 6.2 was evaluated using a parallel artificial membrane permeability assay (PAMPA) evolution instrument (Pion Inc., Billerica, MA) according to the previously published method (Ochi et al., 2022). The bioanalysis was conducted by liquid chromatography–tandem mass spectrometry (LC-MS/MS) (Supplemental Table 1A).
Plasma Protein BindingThe plasma protein binding of TP0473292 in monkeys and humans was evaluated using an ultracentrifugation method. TP0473292 DMSO solution was spiked with monkey or human plasma (1:199, v/v) at a final concentration of 0.1 or 1 μg/ml. After 5-minute incubation at 37°C, an aliquot of the plasma sample was obtained for determining the concentration before ultracentrifugation. The remaining plasma sample was then ultracentrifuged at 627,379g for 4 hours at 10°C. After ultracentrifugation, an aliquot of the plasma supernatant was extracted using acetonitrile/methanol (9:1, v/v) containing the stable isotope-labeled TP0473292. The aliquot of the plasma sample obtained before ultracentrifugation was prepared to have the same compositions as that of the plasma supernatant samples. For the adsorption evaluation, a blank plasma supernatant, which was obtained by the ultracentrifugation of a blank plasma sample at 633,588g for 4 hours at 20°C, was spiked with TP0473292 DMSO solution (1:199, v/v) at a final concentration of 0.1 or 1 μg/ml and incubated for 4 hours at 10°C in the ultracentrifugation tube. The adsorption sample was prepared to have the same compositions as that of the plasma supernatant samples. Each sample was centrifuged at 3974g for 10 minutes at 4°C, and the resultant supernatant was subjected to LC-MS/MS analysis (Supplemental Table 1A). The stability of TP0473292 during ultracentrifugation was investigated, and TP0473292 was confirmed to be stable (>94% remaining after 4 hours of incubation at 10°C). The adsorption ratio (Fads) and the percent of plasma protein binding were calculated using the following equations:

 where C1 is the theoretical initial concentration of TP0473292 in the adsorption sample, C2 is the concentration of TP0473292 in the adsorption sample, Csup is the concentration of TP0473292 in the plasma supernatant sample, and Cp is the concentration of TP0473292 in the plasma sample before ultracentrifugation. The plasma protein binding of TP0178894 was evaluated using an equilibrium dialysis method with a 96-well equilibrium dialysis plate (HTDialysis LLC, Gales Ferry, CT) and a 12–14 kDa cutoff dialysis membrane. TP0178894 was dissolved in DMSO and spiked with rat, monkey, or human plasma (1:199, v/v) at final concentrations of 0.1 and 10 μg/ml (rat and monkey) or 0.1 and 1 μg/ml (human). Each plasma sample was placed in the donor compartment of a 96‐well equilibrium dialysis plate, and sodium phosphate buffer (50 mM, pH 7.4) containing sodium chloride (70 mM) was placed in the reservoir compartment. After incubation for 6 hours at a rate of 100 rpm at 37°C in humidified air (5% CO2), an aliquot from each side of each well was collected and mixed with acetonitrile/methanol (9:1, v/v) containing the stable isotope-labeled TP0178894. Each sample was centrifuged at 3974g for 10 minutes at 4°C, and the resultant supernatant was subjected to LC-MS/MS analysis (Supplemental Table 1A). The protein binding (%) was calculated using the Boudinot formula (Boudinot and Jusko, 1984). The unbound fraction (%) was calculated as 100 – protein binding (%).
where C1 is the theoretical initial concentration of TP0473292 in the adsorption sample, C2 is the concentration of TP0473292 in the adsorption sample, Csup is the concentration of TP0473292 in the plasma supernatant sample, and Cp is the concentration of TP0473292 in the plasma sample before ultracentrifugation. The plasma protein binding of TP0178894 was evaluated using an equilibrium dialysis method with a 96-well equilibrium dialysis plate (HTDialysis LLC, Gales Ferry, CT) and a 12–14 kDa cutoff dialysis membrane. TP0178894 was dissolved in DMSO and spiked with rat, monkey, or human plasma (1:199, v/v) at final concentrations of 0.1 and 10 μg/ml (rat and monkey) or 0.1 and 1 μg/ml (human). Each plasma sample was placed in the donor compartment of a 96‐well equilibrium dialysis plate, and sodium phosphate buffer (50 mM, pH 7.4) containing sodium chloride (70 mM) was placed in the reservoir compartment. After incubation for 6 hours at a rate of 100 rpm at 37°C in humidified air (5% CO2), an aliquot from each side of each well was collected and mixed with acetonitrile/methanol (9:1, v/v) containing the stable isotope-labeled TP0178894. Each sample was centrifuged at 3974g for 10 minutes at 4°C, and the resultant supernatant was subjected to LC-MS/MS analysis (Supplemental Table 1A). The protein binding (%) was calculated using the Boudinot formula (Boudinot and Jusko, 1984). The unbound fraction (%) was calculated as 100 – protein binding (%).
Rat, monkey, or human serum was prewarmed at 37°C, and TP0473292 DMSO solution was added at a final concentration of 50 μM. The rat, monkey, or human intestine, liver, kidney, or lung S9 fraction (0.001–0.1 mg protein/ml) in 250 mM sodium potassium phosphate buffer (pH 7.4), containing 74 mM potassium chloride, 2.4 mM magnesium chloride (MgCl2), 1.4 mM glucose-6-phosphate, and 0.15 mM β-nicotinamide-adenine dinucleotide phosphate oxidized form, was prewarmed at 37°C, and then TP0473292 DMSO solution was added at a final concentration of 10 μM. For the enzyme kinetic studies using the human intestinal or liver S9 fraction, the protein concentration of 0.005 or 0.25 mg/ml and the TP0473292 concentration ranges of 10–500 or 10–1000 μM were used, respectively. The final concentration of DMSO in each incubation mixture was 1% or 1.2% v/v. After incubation for the designated times (1–60 minutes) at 37°C, the reaction was terminated by adding two volumes of acetonitrile/methanol (9:1, v/v) containing the stable isotope-labeled TP0178894, and the resultant mixture was centrifuged at 3974g for 10 minutes at 4°C. An aliquot of the supernatant was subjected to LC‐MS/MS analysis to determine the concentration of TP0178894 (Supplemental Table 1A). The initial rate of TP0178894 formation was calculated as follows:

The kinetic parameters of TP0473292 hydrolysis to TP0178894 in the human intestinal and liver S9 fractions were calculated using Phoenix WinNonlin (version 6.2 or higher; Certara, Princeton, NJ) using the traditional Michaelis-Menten equation and the substrate inhibition model as described below (Tachibana et al., 2005), respectively.
 where V0 is the initial rate of TP0178894 formation (nmol/min per mg protein), Km is the kinetic constant (μM), Vmax is the maximum rate of TP0178894 formation (nmol/min per mg protein), [S] is the substrate (TP0473292) concentration (μM), and Ksi is the substrate inhibition constant (μM).
where V0 is the initial rate of TP0178894 formation (nmol/min per mg protein), Km is the kinetic constant (μM), Vmax is the maximum rate of TP0178894 formation (nmol/min per mg protein), [S] is the substrate (TP0473292) concentration (μM), and Ksi is the substrate inhibition constant (μM).
The effects of the esterase inhibitors, benzil, BNPP, ethephon, EDTA, and PMSF on the hydrolysis of TP0473292 (50 μM) to TP0178894 in the human liver S9 fraction were evaluated. The human liver S9 fraction was preincubated with each esterase inhibitor for 30 minutes on ice before starting the reaction. The reactions were performed as described in the hydrolysis study for the tissue S9 fractions. The final concentration of the human liver S9 fraction in the reaction mixture was 0.25 mg protein/ml and that of the organic solvent was 1.4% v/v. The final concentration of each esterase inhibitor in the reaction mixture was set at a concentration that would sufficiently inhibit the target enzymes based on the previous reports (Table 1). The percent inhibition was calculated as follows:
 where Ccontrol is the concentration of TP0178894 in the absence of the inhibitor, and Cinhibited is the concentration of TP0178894 in the presence of the inhibitor.
where Ccontrol is the concentration of TP0178894 in the absence of the inhibitor, and Cinhibited is the concentration of TP0178894 in the presence of the inhibitor.
Inhibitory effects of esterase inhibitors on the hydrolysis of TP0473292 to TP0178894 in the human liver S9 fraction
Values are presented as the mean of triplicate determinations.
Metabolic Study in Recombinant Human CESThe reaction mixture, consisting of 75 μg/ml of recombinant human CES1 or CES2 and 49 mM phosphate buffer (pH 7.4), was prewarmed at 37°C. The reaction was initiated by adding TP0473292 DMSO solution (2.0% v/v) at a final concentration of 50 μM. After incubation for the designated times (1–15 minutes), the reaction was terminated by adding two volumes of acetonitrile/methanol (9:1, v/v) containing the stable isotope-labeled TP0178894. The resultant mixture was centrifuged at 2150g for 10 minutes at 4°C, and an aliquot of the supernatant was subjected to LC-MS/MS analysis (Supplemental Table 1A). The initial rate of TP0473292 hydrolysis to TP0178894 was calculated as described in the hydrolysis study for the tissue S9 fractions.
Metabolite Profiling of TP0473292 in Rat, Monkey, and Human HepatocytesRat, monkey, or human cryopreserved hepatocytes suspended in Leivovitz L-15 medium containing 2 mM L-glutamine (0.5 million cells/ml) were incubated at 37°C for 1 hour with [3H]TP0473292 and/or TP0473292 at 5 μM (0.1 MBq/ml). The reaction was terminated by adding acetonitrile containing acetic acid (0.1% v/v), and the resultant mixture was centrifuged at 2607g for 10 minutes at 4°C. Aliquots of the supernatant were analyzed using a high-performance liquid chromatograph equipped with a radiochemical flow detector. Aliquots of the supernatant from unlabeled incubation samples and authentic standards were also analyzed using liquid chromatography-mass spectrometry under the same chromatographic conditions, and their chromatographic retention times and mass spectral data were compared (Supplemental Table 1B).
Chemical Stability of Acyl GlucuronideTo evaluate the chemical stability of acyl glucuronide, the degradation rate of acyl glucuronide in the phosphate buffer was evaluated by two incubations (Jinno et al., 2013). First, the test compound (ACA, diclofenac, or ibuprofen) was incubated at 100 µM with pooled human liver microsomes (1 mg protein/ml) in 50 mM phosphate buffer (pH 7.4) containing 4.9 mM MgCl2, 30 μg/ml alamethicin, and 2 mM saccharic acid 1,4-lactone to allow for the formation of acyl glucuronide. Reactions were initiated by adding uridine diphosphate glucuronic acid (5 mM) after 5-minute preincubation at 37°C. After 60-minute incubation, the reactions were terminated by adding 10% v/v acetic acid in acetonitrile/distilled water (1:1, v/v), and the resultant samples were subjected to a second incubation. Second, the aliquots of the resultant samples were spiked into 49 volumes of 200 mM phosphate buffer (pH 7.4) containing 5 mM MgCl2 and further incubated for 0, 0.25, 0.5, 1, 2, and 4 hours to assess the stability of acyl glucuronide in the phosphate buffer. The reactions were terminated by adding 10% v/v acetic acid in acetonitrile/distilled water (1:1, v/v), and the resultant mixtures were subjected to LC-MS/MS analysis after centrifugation (Supplemental Table 1C). The degradation rate constant (k) was determined from the peak responses of acyl glucuronide versus the time curve using linear regression of the semilogarithmic plot, and the half-life (t1/2) was calculated as follows: t1/2 = ln(2)/k.
DDI PotentialP450 Reversible InhibitionThe inhibition potentials of TP0473292, TP0178894, ACA, and ACA-AG on the specific activities of seven human P450 isoforms were evaluated using enzyme specific substrates, phenacetin (CYP1A2), bupropion (CYP2B6), amodiaquine (CYP2C8), diclofenac (CYP2C9), (S)-mephenytoin (CYP2C19), bufuralol (CYP2D6), and midazolam and testosterone (CYP3A), according to a previously published method (Kinoshita et al., 2019). Briefly, the test compound was spiked into pooled human liver microsomes (0.1 mg/ml) at final concentrations of 0.1, 1, or 10 μM and prewarmed. After incubation with a marker substrate of each P450 isoform at a concentration near its Km value (Michaelis constant), the reaction was terminated by adding acetonitrile. The concentration of the specific metabolite of each marker substrate was determined using the previously published LC-MS/MS method (Kinoshita et al., 2019).
P450 Time-Dependent InhibitionThe time-dependent inhibition potentials of TP0473292, TP0178894, ACA, and ACA-AG on the specific activities of seven human P450 isoforms were investigated using the same probe substrates as those used in the reversible inhibition study as well as pooled human liver microsomes according to a previously published method (Takano et al., 2021). Briefly, human liver microsomes (0.5 mg protein/ml) were incubated with the test compound (10 μM) for 30 minutes at 37°C to allow for the generation of intermediates that may inhibit P450 isoforms. Then, the first incubation mixture was diluted 10-fold with the second incubation mixture containing each probe substrate at a final concentration near fivefold of its Km value. The second incubation was conducted for the designated time at 37°C and was terminated by adding acetonitrile. The concentration of the specific metabolite of each marker substrate in the second incubation mixture was determined using the previously reported LC-MS/MS method (Kinoshita et al., 2019).
P450 InductionThe CYP1A2, CYP2B6, and CYP3A4 induction potentials of TP0473292 and TP0178894 (0.1, 1, and 10 μM) were evaluated using mRNA levels in the primary cultured cryopreserved human hepatocytes according to a previously published method (Takano et al., 2021), with some modifications. The mRNA expression levels of CYP1A2, CYP2B6, and CYP3A4 were measured using a fluorescent-labeled microbead assay using the QuantiGene Plex 2.0 Plex Set (customized product of panel no. 12117) and the QuantiGene Plex 2.0 Assay Kit. The CYP1A2, CYP2B6, and CYP3A4 mRNA levels were normalized against the endogenous control gene (β-glucuronidase).
Pharmacokinetics in Rats and MonkeysThe pharmacokinetics of TP0473292 and TP0178894 in fasted male SD rats (n = 3) and fasted male cynomolgus monkeys (n = 3) were investigated. For intravenous administration, TP0178894 was dissolved in saline and administered to rats and monkeys (1 mg/kg). TP0473292 was dissolved in 10% w/v 2-hydroxypropyl-β-cyclodextrin containing 2.5% w/v mannitol and administered to rats and monkeys (1 mg/kg, 0.613-mg equivalent of TP0178894/kg). Blood samples were collected from the tail vein of rats or the cephalic vein of monkeys into tubes containing the anticoagulant EDTA-2K (rats and monkeys) and the esterase inhibitor PMSF (rats only) at predose (monkeys only) and 5, 15, and 30 minutes and 1, 2, 4, 8, and 24 hours postdose, and then plasma samples were obtained after centrifugation. Urine samples were collected over the intervals of 0–24 hours (rats) or 0–8 and 8–24 hours (monkeys) into bottles containing 20% v/v acetic acid on ice. For oral administration, TP0178894 was suspended in 0.5% w/v methylcellulose 400 and administered to monkeys (1 mg/kg). TP0473292 was suspended in 0.5% w/v methylcellulose 400 and administered to rats (1 mg/kg, 0.613-mg equivalent of TP0178894/kg) and monkeys (1.63 mg/kg, 1-mg equivalent of TP0178894/kg). Blood samples were collected from the tail vein of rats or the cephalic vein of monkeys into tubes containing EDTA-2K (rats and monkeys) and PMSF (rats only) at 30 minutes and 1, 2, 4, 8, and 24 hours postdose (rats) or at predose, 30 minutes, and 1, 2, 3, 4, 6, 8, and 24 hours postdose (monkeys). The plasma or urine samples were precipitated with acetonitrile/methanol (9:1, v/v) containing the stable isotope-labeled TP0178894 or a mixture of the stable isotope-labeled TP0473292, TP0178894, ACA, and ACA-AG. Following centrifugation at 3639g for 10 minutes at 4°C, the aliquots of the supernatant were analyzed by LC‐MS/MS (Supplemental Table 1, A and D). To evaluate the extent of the cerebrospinal fluid (CSF) penetration of TP0178894 in rats, TP0473292 was suspended in 0.5% w/v methylcellulose 400 and administered orally to nonfasted SD rats at a dose of 1 mg/kg (0.613-mg equivalent of TP0178894/kg). Blood samples were collected from the posterior vena cava into tubes containing EDTA-2K and PMSF under isoflurane anesthesia at 1, 2, 3, 4, 8, or 24 hours postdose. Then, the animal was euthanized by exsanguination, and CSF was drawn from the spinal cord space. The plasma or CSF was precipitated with acetonitrile/methanol (9:1, v/v) containing TP0181164, a structural analog of TP0178894, as an internal standard and centrifuged. Aliquots of the supernatant were analyzed by LC‐MS/MS (Supplemental Table 1A).
Pharmacokinetic AnalysisThe concentration-time profiles of TP0473292, TP0178894, ACA, and ACA-AG in plasma were analyzed using a noncompartmental analysis (Phoenix WinNonlin version 6.2).
ResultsPAMPA PermeabilityThe membrane permeability of TP0473292 and TP0178894 at pH 6.2 was evaluated using a PAMPA at 10 µM. The apparent permeability of TP0473292 was 36.6 × 10−6 cm/s, whereas the apparent permeability of TP0178894 was estimated to be less than 0.03 × 10−6 cm/s since the concentrations in the acceptor solutions were below the lower limit of quantification (0.006 µM).
Plasma Protein BindingThe unbound fractions of TP0473292 at 0.1 and 1 μg/ml ranged from 1.7% to 1.8% in monkeys and 1.1% in humans. The protein binding of TP0473292 in rat plasma was not evaluated, because of its rapid hydrolysis to TP0178894. The unbound fractions of TP0178894 in rat, monkey, and human plasma (0.1 and 10 μg/ml in rats and monkeys, 0.1 and 1 μg/ml in humans) ranged from 87.6% to 87.9%, 100.4% to 101.0%, and 92.6% to 94.9%, respectively. These data indicate that there are no apparent species differences or concentration dependencies in plasma protein binding for TP0473292 or TP0178894.
Hydrolysis of TP0473292 to TP0178894 in Rat, Monkey, and Human Sera and Tissue S9 FractionsThe formation rates of TP0178894 from TP0473292 in rat, monkey, and human sera (50 µM TP0473292) and tissue S9 fractions (10 μM TP0473292) are shown in Table 2. The formation rate of TP0178894 in serum was much faster in rats than that in monkeys and humans, with a TP0473292 elimination t1/2 of less than 1 hour, assuming complete conversion to TP0178894. In rats, the formation rate of TP0178894 was highest in the intestinal S9 fraction, followed by the lung, liver, and kidney S9 fractions. In monkeys, the formation rate of TP0178894 was higher in the intestinal and kidney S9 fractions than in the liver and lung S9 fractions. In humans, the formation rate of TP0178894 was higher in the liver and intestinal S9 fractions than the kidney and lung S9 fractions. The enzyme kinetics for the hydrolysis of TP0473292 to TP0178894 in human intestinal and liver S9 fractions were evaluated. The formation rates of TP0178894 ranged from 4.25 to 19.6 nmol/min per mg protein in the human intestinal S9 fraction for TP0473292 concentration ranges of 10–500 µM and from 15.9 to 204 nmol/min per mg protein in the liver S9 fraction for TP0473292 concentration ranges of 10–1000 µM. The Eadie-Hofstee plots showed that the hydrolysis of TP0473292 to TP0178894 exhibited Michaelis-Menten kinetics in the human intestinal S9 fraction and substrate inhibition kinetics in the human liver S9 fraction (Supplemental Fig. 1). The Km in the human intestinal S9 fraction was calculated to be 40.3 μM, and the Vmax was 21.3 nmol/min per mg protein. The Km and Ksi in the human liver S9 fraction were calculated to be 583 μM and 141 μM, respectively, and the Vmax was 927 nmol/min per mg protein. The Km value in the human intestinal S9 fraction was comparable to its solubility in the fed-state simulated intestinal fluid (data not shown), the assumed highest concentration in the gut. The Km and Ksi in the human liver S9 fraction were sufficiently higher than the plasma concentration at an effective dose in rat models (approximately 2 μM as TP0178894) and probably in humans as well since no species difference in antagonist activity was observed between rats and humans (Watanabe et al., 2022). These results indicated that TP0473292 hydrolysis would not be saturated in either the intestine or the liver at an effective dose in humans.
TABLE 2Formation rate of TP0178894 from TP0473292 in rat, monkey, and human sera (50 µM TP0473292) and tissue S9 fractions (10 µM TP0473292)
Values are presented as the mean (S.D.) of triplicate determinations.
Reaction PhenotypingChemical Inhibition StudyThe esterases involved in the hydrolysis of TP0473292 to TP0178894 in the human liver S9 fraction were estimated by selective chemical inhibition studies with five esterase inhibitors: benzil, BNPP, ethephon, EDTA, and PMSF. The inhibitory effects of these esterase inhibitors are shown in Table 1. The paraoxonase inhibitor EDTA and the butyrylcholinesterase inhibitor ethephon showed minimal inhibition of the hydrolysis of TP0473292 to TP0178894, indicating that neither paraoxonase nor butyrylcholinesterase was likely to be involved in the hydrolysis of TP0473292. BNPP and PMSF inhibited the hydrolysis of TP0473292 to TP0178894 completely, suggesting that CES and/or arylacetamide deacetylase were involved in the hydrolysis of TP0473292. Benzil, which inhibits CES but not arylacetamide deacetylase (Shimizu et al., 2014), also inhibited the hydrolysis of TP0473292 completely, suggesting that CES, but not arylacetamide deacetylase, was responsible for the hydrolysis of TP0473292. As CES4 is less sensitive to PMSF inhibition (Fujiyama et al., 2009), among CES1, CES2, and CES4 expressed in human liver, CES1 and/or CES2 were likely to be responsible for the hydrolysis of TP0473292 to TP0178894 in the human liver S9 fraction.
Metabolic Study in Recombinant Human CESThe formation rates of TP0178894 from TP0473292 (50 µM) in recombinant human CES1 and CES2 were 7.4 and 19.8 nmol/min per mg protein, respectively. These results indicated that the hydrolysis of TP0473292 to TP0178894 is mediated by both CES1 and, more efficiently, CES2.
Metabolite Profiling of TP0473292 in Rat, Monkey, and Human HepatocytesThe metabolite profiles of TP0473292 in rat, monkey, and human cryopreserved hepatocytes were evaluated. In all the species, the predominant peak was identified as TP0178894 based on the mass spectral data and chromatographic retention time, and no other metabolite peak was detected by the high-performance liquid chromatograph equipped with the radiochemical flow detector (Fig. 2). In addition to TP0178894, the mass chromatograms detected ACA and ACA-AG in all the species (Supplemental Table 2). The proposed metabolic pathways of TP0473292 in rats, monkeys, and humans are shown in Fig. 1B. TP0473292 underwent enzymatic hydrolysis to yield TP0178894 and ACA, and ACA was further metabolized via glucuronidation to form ACA-AG. The further metabolism of TP0178894 was not observed, and no human-specific metabolites were found.
 Fig. 2.
Fig. 2. HPLC radiochromatograms of the incubation mixtures of [3H]TP0473292 with rat (A), monkey (B), and human (C) cryopreserved hepatocytes. [3H]TP0473292 (5 μM) was incubated with rat, monkey, or human cryopreserved hepatocytes at 0.5 million cells/ml at 37°C for 1 hour.
Chemical Stability of Acyl GlucuronideThe chemical stability of ACA-AG was evaluated in phosphate buffer (pH 7.4). ACA-AG showed a much longer degradation t1/2 (79.2 hours) compared with the acyl glucuronides of diclofenac (1.23 hours) and ibuprofen (4.92 hours), which are classified as “warning” in terms of their idiosyncratic drug toxicity (IDT) risk (Sawamura et al., 2010).
DDI PotentialP450 InhibitionThe inhibitory potentials of TP0473292, TP0178894, ACA, and ACA-AG on seven human P450 isoforms, CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A, were investigated using human liver microsomes. All the compounds had no inhibitory potential for any of the P450 isoforms at concentrations up to 10 µM in a reversible or a time-dependent manner (Supplemental Table 3).
P450 InductionThe induction potentials of TP0473292 and TP0178894 on CYP1A2, CYP2B6, and CYP3A4 mRNA expressions were evaluated using three lots of primary cultured cryopreserved human hepatocytes at a concentration of 0.1, 1, or 10 μM. TP0473292 or TP0178894 did not produce marked changes in CYP1A2, CYP2B6, or CYP3A4 mRNA levels at concentrations up to 10 µM (Supplemental Table 4). Since ACA and ACA-AG were probably formed during the TP0473292 induction study, both metabolites were thought to have no apparent induction potential on the tested P450 mRNA.
Pharmacokinetics of TP0178894 in Rats and MonkeysThe plasma concentration-time profiles of TP0178894 after a single intravenous administration of TP0178894 (1 mg/kg) to male rats and monkeys and that after a single oral administration of TP0178894 (1 mg/kg) to male monkeys under fasted conditions are shown in Fig. 3. The pharmacokinetic parameters are summarized in Table 3. After the intravenous administration of TP0178894, the plasma TP0178894 level rapidly decreased, with an elimination t1/2 of 1.08 hours in rats and 1.82 hours in monkeys. The volume of distribution at steady state was low: 473 ml/kg in rats and 258 ml/kg in monkeys. The excretion of TP0178894 into urine within 24 hours postdose was 76.6% of the dose in rats and 97.4% of the dose in monkeys. The renal clearance was calculated to be 403 ml/h per kg in rats and 263 ml/h per kg in monkeys. After the oral administration of TP0178894 to monkeys, TP0178894 slowly reached a plasma Cmax of 22.6 ng/ml at 3.50 hours postdose and then slowly declined with a t1/2 of 20.0 hours. The oral bioavailability was estimated to be 5.8%.
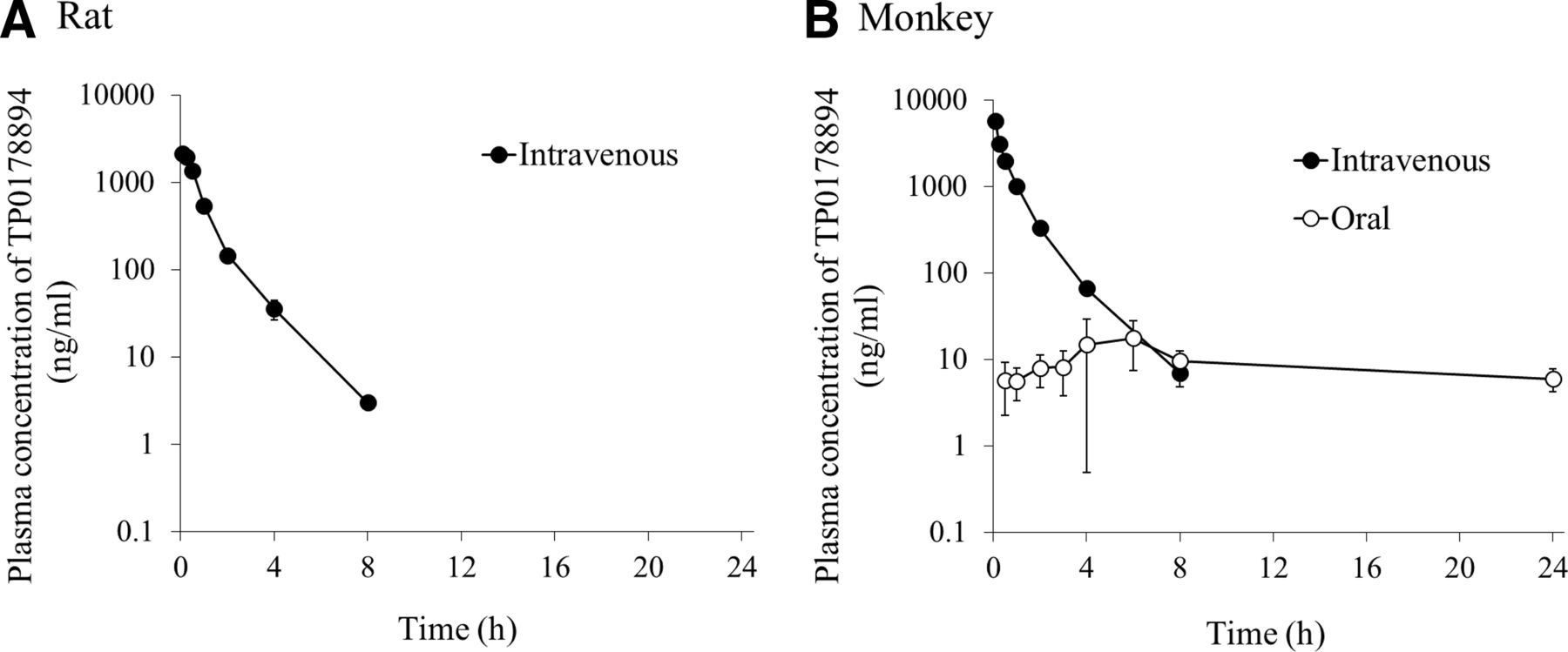 Fig. 3.
Fig. 3. Plasma concentration-time profiles of TP0178894 after a single intravenous or oral administration of TP0178894 to fasted male SD rats (A) or cynomolgus monkeys (B) at a dose of 1 mg/kg. Data are represented as the mean ± S.D. of three animals. The lower limit of quantification was 1 ng/ml for rats and 0.5 ng/ml for monkeys.
TABLE 3Pharmacokinetic parameters of TP0178894 after a single intravenous (fasted male SD rats and fasted male cynomolgus monkeys) or oral (fasted male cynomolgus monkeys) administration of TP0178894 at a dose of 1 mg/kg
Data are presented as the mean (S.D.) of three animals.
Pharmacokinetics of TP0473292 in Rats and MonkeysThe plasma concentration-time profiles of TP0473292, TP0178894, ACA, and ACA-AG after a single intravenous or oral administration of TP0473292 to male rats (1 mg/kg, 0.613-mg equivalent of TP0178894/kg) and monkeys (1 or 1.63 mg/kg, 0.613- or 1-mg equivalent of TP0178894/kg, respectively) under fasted conditions are shown in Fig. 4 and Fig. 5, respectively. The pharmacokinetic parameters are summarized in Table 4 and Table 5, respectively. After the intravenous administration of TP0473292 to rats and monkeys, TP0473292 rapidly declined with a t1/2 of 0.375–0.624 hours. TP0178894 was detected from the first sampling time point (5 minutes postdose), reaching a Cmax at 0.500–0.917 hours postdose, followed by a gradual decline (t1/2 = 1.09–3.54 hours), compared with TP0473292. The plasma levels of ACA, liberated together with TP0178894 from TP0473292, and its acyl glucuronide, ACA-AG, immediately reached their Cmax within 0.25 hours postdose and declined with a t1/2 of less than 3 hours. The urinary excretion of TP0178894 and ACA-AG within 24 hours postdose was 80.3% and 65.6% of the administered dose in rats and 85.7% and 71.1% in monkeys, respectively. The excretion of TP0473292 and ACA into urine was less than 1.5% of the administered dose in both species. After the oral administration of TP0473292 to rats and monkeys, TP0178894 was detected from the first sampling time point (30 minutes postdose) and reached a Cmax at 1.00–2.67 hours postdose. TP0178894 declined with a t1/2 of 1.44–4.68 hours, which was slightly slower than that after intravenous administration. TP0473292 was not detected in plasma in both animals. The oral bioavailability of TP0178894 after TP0473292 administration was 58.4% in rats and 56.6% in monkeys, which was approximately 10-fold higher than that after the oral administration of TP0178894 itself in monkeys (5.8%). The plasma levels of ACA and ACA-AG immediately reached their Cmax within 2 hours postdose and declined with a t1/2 of approximately 4 to 5 hours, which was slower than that after intravenous administration. The extent of CSF penetration of TP0178894 after a single oral administration of TP0473292 was evaluated in nonfasted male rats at a dose of 1 mg/kg (Fig. 6; Table 6); at this dose, the immobility time in the rat forced-swimming test was previously reported to be significantly reduced (Watanabe et al., 2022). The CSF concentration of TP0178894 reached a Cmax at 3 hours postdose, which was later than that in plasma (1 hour) and decreased at a slower rate than that in plasma. The area under the concentration-time curve (AUC) from 0 to 8 hours of CSF was 1.5% of that in plasma.
 Fig. 4.
Fig. 4. Plasma concentration-time profiles of TP0473292, TP0178894, ACA, and ACA-AG after a single intravenous (A) or oral (B) administration of TP0473292 to fasted male SD rats at a dose of 1 mg/kg. Data are represented as the mean ± S.D. of three animals. The lower limits of quantification for TP0473292, TP0178894, ACA, and ACA-AG were 1, 1, 3, and 1 ng/ml, respectively. Plasma TP0473292 concentrations were below the lower limit of quantification at all time points after a single oral administration of TP0473292.
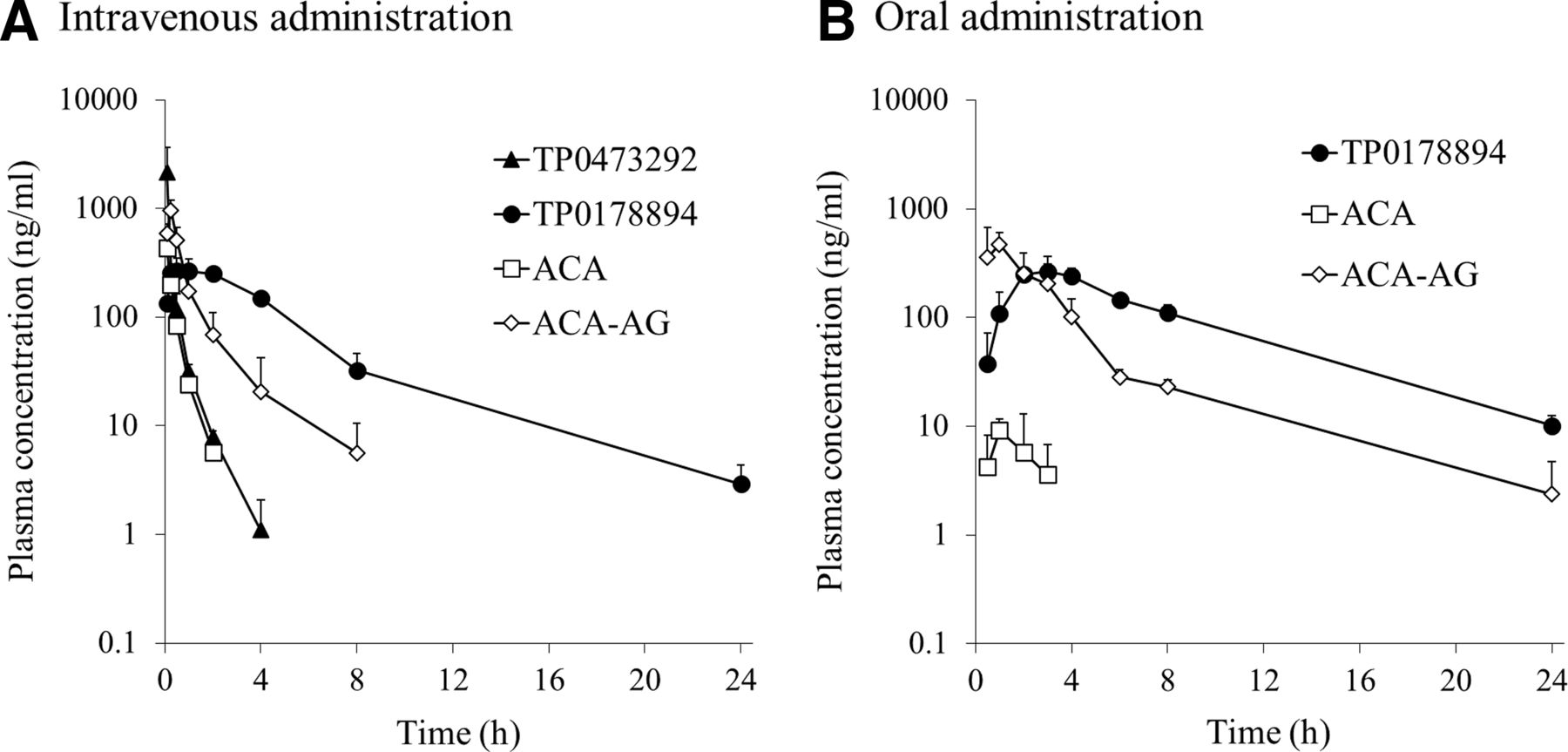 Fig. 5.
Fig. 5. Plasma concentration-time profiles of TP0473292, TP0178894, ACA, and ACA-AG after a single intravenous [(A) 1 mg/kg] or oral [(B) 1.63 mg/kg] administration of TP0473292 to fasted male cynomolgus monkeys. Data are represented as the mean ± S.D. of three animals. The lower limits of quantification for TP0473292, TP0178894, ACA, and ACA-AG were 1, 1, 3, and 1 ng/ml, respectively. Plasma TP0473292 concentrations were below the lower limit of quantification at all time points after a single oral administration of TP0473292.
 Fig. 6.
Fig. 6. Plasma and CSF concentration-time profiles of TP0178894 after a single oral administration of TP0473292 to nonfasted male SD rats at a dose of 1 mg/kg. Data are represented as the mean ± S.D. of three animals at each time point. The lower limit of quantification for TP0178894 was 0.1 ng/ml in plasma and 0.2 ng/ml in CSF.
TABLE 4Pharmacokinetic parameters of TP0473292, TP0178894, ACA, and ACA-AG after a single intravenous or oral administration of TP0473292 to fasted male SD rats at 1 mg/kg
Data are presented as the mean (S.D.) of three animals.
TABLE 5Pharmacokinetic parameters of TP0473292, TP0178894, ACA, and ACA-AG after a single intravenous (1 mg/kg) or oral (1.63 mg/kg) administration of TP0473292 to fasted male cynomolgus monkeys
Data are presented as the mean (S.D.) of three animals.
TABLE 6Pharmacokinetic parameters of TP0178894 in plasma and CSF after a single oral administration of TP0473292 to nonfasted male SD rats at a dose of 1 mg/kg
Parameters were calculated from the mean concentration of three animal
留言 (0)