
記住我

Patients demonstrating ≥ 2 signs and symptoms and with a confirmed genetic diagnosis of NNS/CANDLE, SAVI, or AGS (including familial chilblain lupus) were eligible. Patients with NNS/CANDLE or SAVI ≥ 17.5 months of age and patients with AGS ≥ 6 months of age were included in the trial [17]. All the patients weighed ≥ 5 kg with average daily dairy score (DDS) of
≥ 0.5 for patients with NNS/CANDLE at Visit 2,
≥ 1.0 for patients with SAVI at Visit 6, and
≥ 0.5 for patients with AGS at Visit 6
Study designThis Phase 2/3, multicenter, open-label study (ClinicalTrials.gov Identifier: NCT04517253) evaluated the efficacy and safety of baricitinib in adult and pediatric Japanese patients with autoinflammatory type I interferonopathies including NNS/CANDLE, SAVI, and AGS.
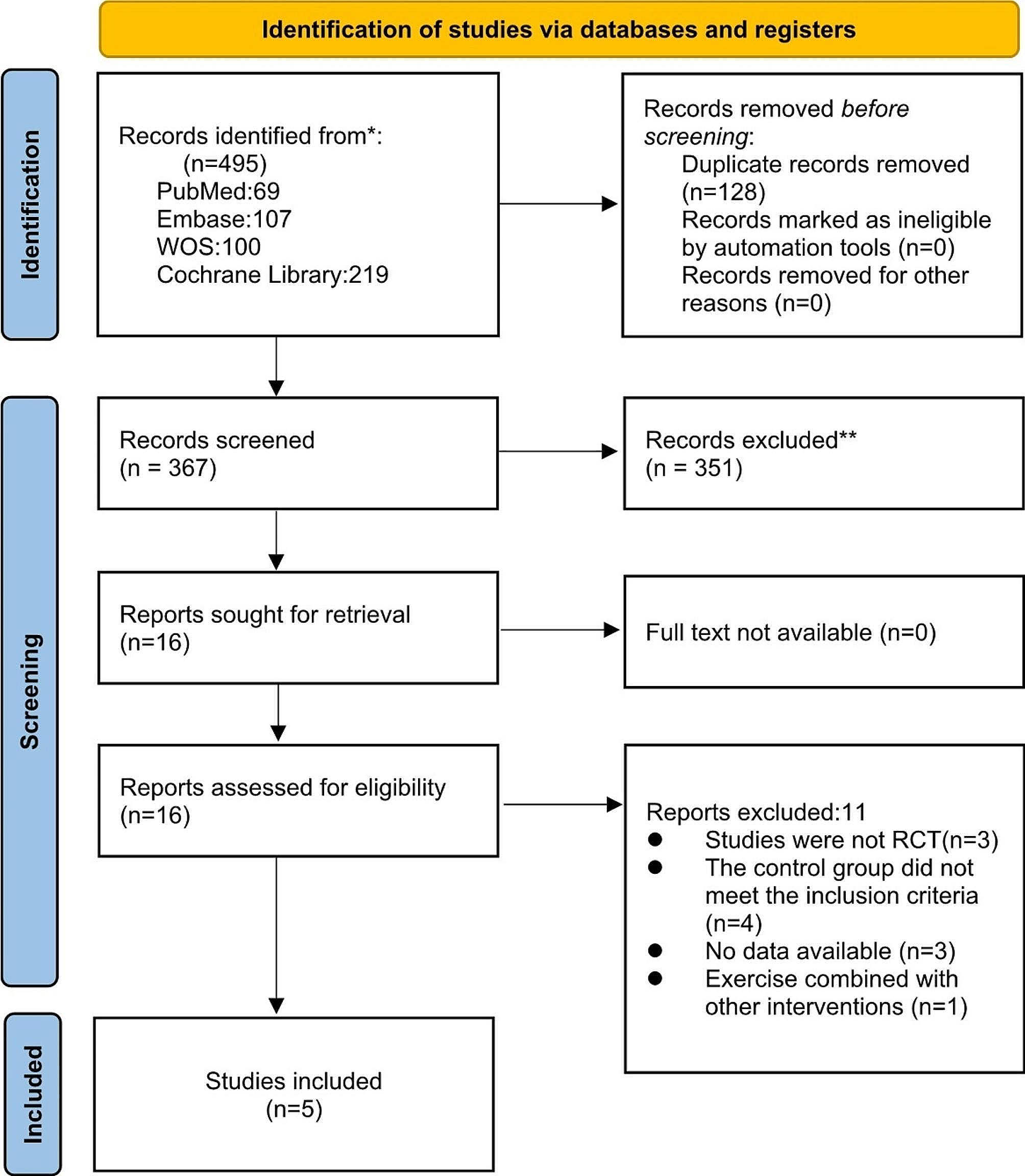
The study was divided into 6 periods including (i) screening, (ii) pre-treatment (only for patients with NNS/CANDLE), (iii) dose adjustment, (iv) primary treatment, (v) maintenance treatment, and (vi) post-treatment follow-up period (Fig. 1).
Fig. 1
Study design. AGS = Aicardi-Goutières syndrome; NNS/CANDLE = Nakajo-Nishimura syndrome/chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature; SAVI = STING-associated vasculopathy with onset during infancy; STING = stimulator of interferon genes; V = visit; wk = week. aPatient can skip Visit 5 and proceed to Visit 6 if biologic agents are not administered during pre-treatment period or an appropriate washout duration of biologic agents defined in Exclusion Criterion #28 has already passed at Visit 5. bVisit 801 should occur approximately 28 days after the last dose of study drug. Patients who will transition from this study to commercial baricitinib are not required to complete Visit 801
The screening period varied for patients with NNS/CANDLE, SAVI, and AGS. The screening period ranged from 7 to 35 days prior to baseline (Visit 6) for patients with SAVI and AGS, whereas patients with NNS/CANDLE had a screening period of 1 week (Visit 1) before entering the pre-treatment period.
Patients with NNS/CANDLE entered a 12-week pre-treatment period at the beginning of Visit 1. The data from the pre-treatment period was used for baseline comparison.
Patients who met all eligibility criteria entered the 8-week dose adjustment period (Visit 6 to Visit 9). Patients initially received treatment dose based on weight class and eGFR, which was further escalated to identify tolerable dose. The dose escalation model was based on the results from previous PK studies of baricitinib [11, 16]. Patients who weighed < 40 kg received baricitinib tablets or liquid suspension orally based on patient choice, whereas patients who weighed ≥ 40 kg were recommended to receive only tablets. If the patients’ weight changed during the study, dose regimens could be modified. Compliance was assessed by counting returned tablets or weighing a returned bottle for liquid suspension. Patients treated with baricitinib were considered noncompliant if they missed ≥ 20% of the prescribed doses during the study or if they were judged by the investigator to have intentionally or repeatedly taken more than the prescribed amount of study medication.
Patients received the optimized dosage throughout the primary and maintenance treatment periods. The duration of primary treatment period was 12 weeks for patients with NNS/CANDLE and 24 weeks for patients with SAVI and AGS based on the results from the JAGA (NCT01724580) trial [11]. Entry into the maintenance period after completion of the primary treatment period occurred at Visit 12 for patients with NNS/CANDLE and Visit 15 for patients with SAVI and AGS. This study is ongoing to collect long-term efficacy and safety results.
Treatment period was considered from the first dose of baricitinib (Visit 6; Week 0) to the date of final visit or early discontinuation period. Endpoint of post-follow-up period was Visit 801. Primary endpoint assessed the change in mean DDS from baseline (Week 0; Visit 6) to Week 20 in patients with NNS/CANDLE and Week 32 in patients with SAVI and AGS. Other efficacy and safety endpoints were assessed until Week 52.
The study protocol was approved by the institutional review boards prior to patient recruitment, and each patient or their guardian provided written informed consent prior to enrollment. The study was conducted in accordance with consensus ethics principles derived from international ethics guidelines, including the Declaration of Helsinki and the International Ethical Guidelines by the Council for International Organizations of Medical Sciences and the International Council for Harmonization E6 Guidelines for Good Clinical Practice.
Clinical benefit assessmentDecrease in daily diary scoreThe primary effectiveness measure evaluated the decrease in DDS of patients’ signs and symptoms. Daily symptoms, including fever, rash, musculoskeletal pain, headache, and fatigue, were rated with increasing level of severity. Signs and symptoms for NNS/CANDLE, SAVI, and AGS are detailed in Supplementary Table 1. The average score of each symptom was calculated using the data from 7 days preceding the current visit. The calculated average score for each symptom is summed up and divided by the number of assessed symptoms (5 symptoms for NNS/CANDLE, 6 symptoms for SAVI, and 8 symptoms for AGS) to calculate the average score for each patient. DDS does not change linearly; therefore, no settings for "minimal clinically important difference” could be set. However, patients were considered well-controlled if a DDS of < 0.5 for NNS/CANDLE and < 1.0 for SAVI was observed [11].
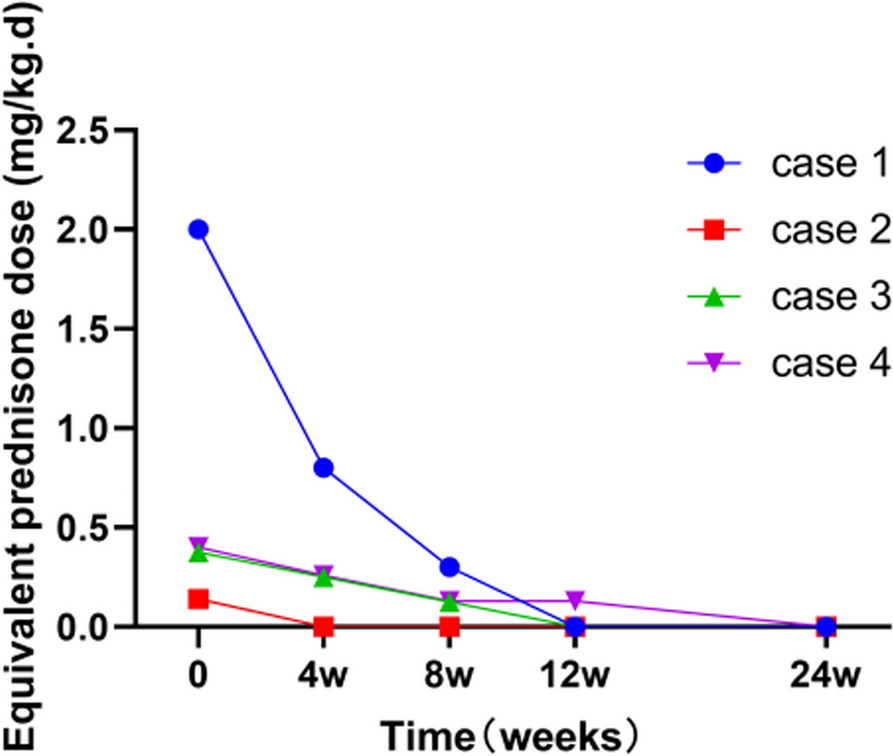
Decrease in daily dose of corticosteroidsA decrease in daily dose of corticosteroids was defined by a < 0.15 mg/kg/day prednisone equivalent systemic corticosteroid dose or a daily dose percentage decrease of at least 50% from baseline.
Decrease in physician’s global assessment of disease activity scoreThe patients’ current disease activities were assessed using the 21-circle visual analog scale ranging from 0 to 10 with increasing level of severity [18].
Other clinical assessmentsAn improvement in diary symptom-specific score (DSSS) from baseline, Barthel index evaluating the activity of living with intractable diseases [19], mean changes in height and growth from baseline, and laboratory parameters including C-reactive protein (CRP), aspartate transaminase, alanine aminotransferase, gamma glutamyl transferase, and creatine phosphokinase were assessed.
Mean change from baseline in biomarkers of IFN signaling and IGS was assessed throughout the study. The IFN signature score used in IGS assessment, comprised of 6-gene IFN signature assays from 28 IFN Response Genes. The final assay was parameterized to separate IFN-high and IFN-low patients with a cut point of zero where each gene was scaled to have the same range. A change of 1 point in the IFN signature means twofold change in gene concentration.
Safety assessmentAll adverse events (AEs) occurring after signing the informed consent form were recorded in the electronic case report form. AEs were classified based on the Medical Dictionary for Regulatory Activities. AE of special interest included infections, myelosuppressive events, thrombocytosis, malignancies, hepatic events, major adverse cardiovascular events, and thrombotic events. Electrocardiograms, physical examinations, vital signs including lung and liver function tests, BK virus plasma and urine screening using quantitative PCR, hepatitis B virus DNA monitoring (only in patients who were positive for HbcAb or HbsAb at Visit 1), and height and weight measurement were performed, and clinically significant findings were reported.
StatisticsIn lieu of anticipating that relatively few patients with each condition would be enrolled, no formal statistical tests were planned. Instead, descriptive summaries and data listings were planned to summarize the results.
Continuous data were summarized in terms of the mean, standard deviation (SD), minimum, maximum, median, and number of observations; categorical data were summarized as frequency counts and percentages. Although the efficacy population set included all enrolled patients who had at least 1 dose of baricitinib, efficacy analyses were divided among diagnosis-specific efficacy population subsets.
Mean DDS was calculated from the average scores of 7 days preceding the current visit; however, if more than 50% symptom scores were missing, the mean DDS was not calculated. A last-observation-carried-forward imputation replaced missing data with the most recent non-missing postbaseline assessment. Proportion of days meeting threshold was calculated by the total number of days meeting the threshold divided by total number of days with non-missing diary scores during the interval. Mean DDS with < 0.5 was used as a threshold for all diseases as an indication of disease control. All corticosteroid doses were standardized to an equivalent prednisone dose.
留言 (0)