Ethical Use of Animals
All mice were maintained by animal care staff and veterinarians of the University of Frankfurt/Main Zentrale Forschungs-Einrichtung (ZFE). All procedures were performed according to the German Animal Welfare Act and the guidelines of the Federation of European Laboratory Animal Science Associations, based on European Union legislation (Directive 2010/63/EU). Animal experiments were approved by the local ethics committee (Regierungs-Präsidium Darmstadt V54-19c18-FK/1083).
Ethical Use of Human Tissue
All the work involving human tissue has been carried out in accordance with the Code of Ethics of the World Medical Association (Declaration of Helsinki) and with national legislation as well as our institutional guidelines. Experiments were approved by the local ethics committee (ethical vote Tübingen, 598/2011BO1 and 911/2019BO2).
Mouse and iPSC Sample Description
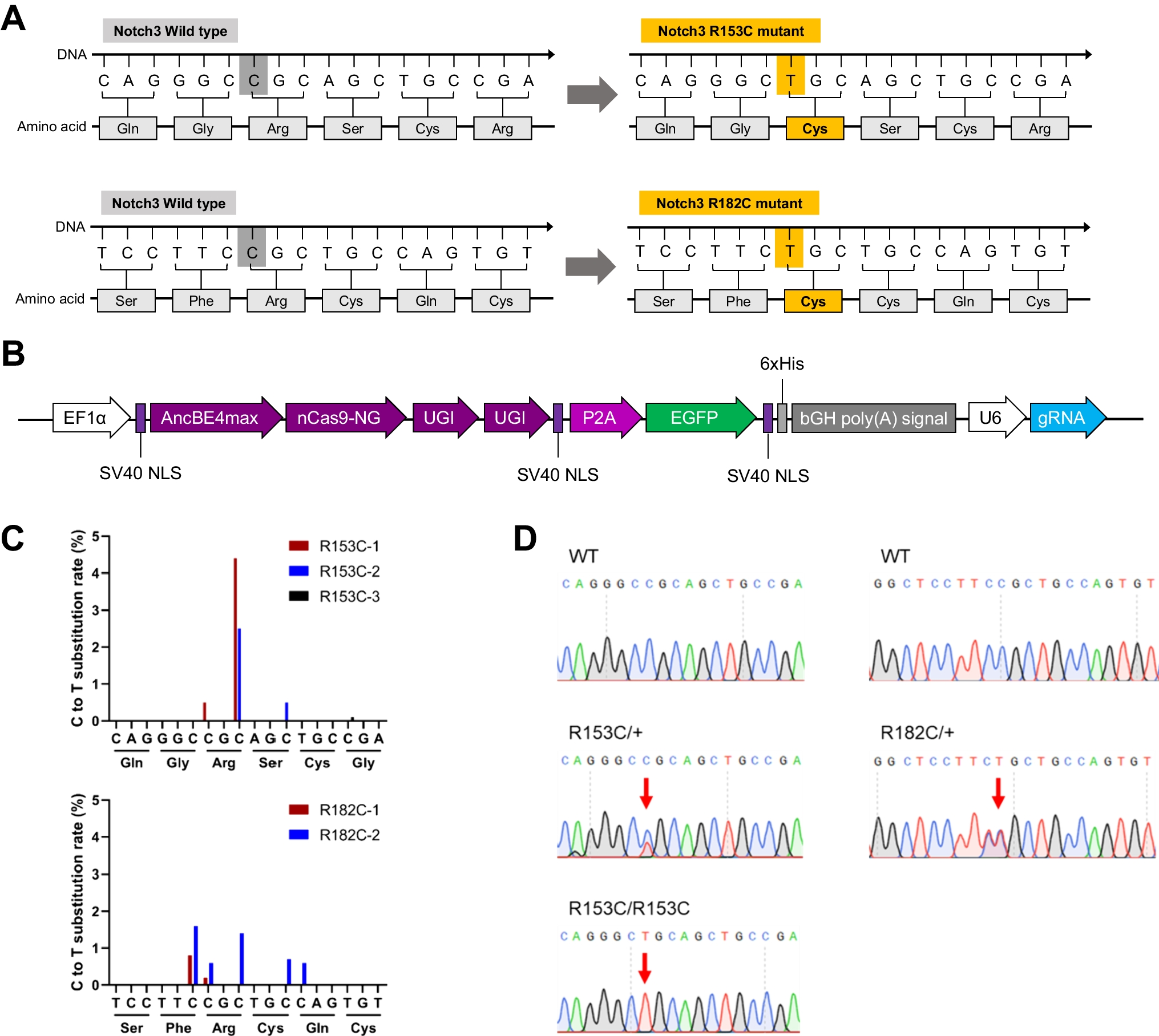
Different mouse tissues (liver, hemisphere, cerebellum) isolated from young (2.5–3 months) and old (14–18 months) knock-in mouse lines modeling SCA2, including ATXN2-CAG42 [58] and Atxn2-CAG100 knock-in mice [59], were used during the establishment of the assay conditions. The generation of mouse embryonic fibroblast (MEF) lines from Atxn2-CAG100 knock-in mice was reported before [59]. Human fibroblasts were isolated from skin biopsies of three SCA2 patients and three neurologically healthy controls after written informed consent. Fibroblasts were reprogrammed into induced pluripotent stem cells (iPSCs) as specified earlier [60] and differentiated into cortical neurons (CNs) as described before [61]. Specification of analyzed human cell lines are represented in Table 1. Human serum samples from four SCA2 patients and five neurologically healthy controls were isolated using serum preparation tubes (BD Biosciences, Heidelberg, Germany).
Table 1 Specification of human cell lines used in this studyCell Culture and Transfection
MEF and human embryonic kidney (HEK293T) cells were maintained in DMEM (Thermo Fisher Scientific, Waltham, USA) supplemented with 10% fetal bovine serum (FBS) and 1% antibiotics-antimycotics (both Thermo Fisher Scientific, Waltham, USA) at 5% CO2 and 37 °C. For MEF, experiments were carried out only within the first 5 passages. Different ATXN2 plasmids including GFP-ATXN2-22Q/myc-ATXN2-22Q, GFP-ATXN2-79Q/myc-ATXN2-79Q, or GFP-empty/myc-empty as internal control were used for transfection experiments.
HEK293T cells were transfected with GFP or myc ATXN2 plasmids with different polyQ length (GFP-22Q/myc-22Q, GFP-79Q/myc-79Q, or GFP-empty/myc-empty as internal control) with Attractene transfection reagent (Qiagen, Hilden, Germany) using the traditional transfection protocol. Shortly, 24 h before transfection, 400,000 HEK293T cells per well were seeded on a six-well tissue culture plate in DMEM medium. For transfection, 1.2 μg plasmid DNA, 100 μl OptiMEM (Thermo Fisher Scientific, Waltham, USA), and 4.5 μl Attractene Transfection Reagent were mixed, incubated for 15 min at room temperature (RT), and afterwards dropped carefully on the seeded cells. Seventy-two hours after transfection, HEK293T cells as well as MEF cells were harvested with DPBS (Thermo Fisher Scientific, Waltham, USA), centrifuged for 5 min at 300g, and directly lysed in RIPA buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% IGEPAL) supplemented with cOmplete (EDTA-free) Protease Inhibitor (Roche, Mannheim, Germany) followed by incubation on ice for 30 min and vortexing every 10 min. For lysate preparation, the cell homogenates were centrifuged for 30 min at 4 °C and 16,100g. The Bradford protein assay was used to determine the total protein concentration [62].
Cell Culture Starvation and siRNA Experiments
For the starvation experiments, 400,000 HEK293T cells per well were seeded on a six-well tissue culture plate and transfected with ATXN2 GFP-22Q, GFP-79Q or GFP-empty as control, as described above.
Seventy-two hours after transfection, the medium was changed to Hanks’ Balanced Salt Solution (Thermo Fisher Scientific, Waltham, USA) and incubated for either 0 h, 1 h, or 2 h, respectively. After incubation, cells were harvested as described.
For esiRNA experiments, knockdown of ATXN2 was achieved using endoribonuclease-prepared siRNA (esiRNA) directed against human ATXN2 (MISSION® esiRNA EHU104101, Sigma-Aldrich, Missouri, USA) or control esiRNA against Renilla luciferase (MISSION® esiRNA EHURLUC, Sigma-Aldrich, Missouri, USA). Four hundred thousand HEK293T cells per well were seeded on a six-well tissue culture plate. The transfection followed the traditional Attractene transfection protocol (Qiagen, Hilden, Germany) with 1.2 μg plasmid DNA (ATXN2 GFP-22Q, ATXN2 GFP-79Q or GFP-empty as control), 18 pmol esiRNA, and 4.5 μl Attractene Transfection Reagent. Seventy-two hours after transfection, cells were harvested with DPBS, centrifuged for 5 min at 300g and lysed in RIPA for 30 min on ice and vortexing every 10 min.
Isolation of Recombinant ATXN2
HEK293T cells (1.2 × 107) were seeded per 175 cm2 cell culture dish. After 24 h, cells were transfected with 8 μg ATXN2 Myc-22Q or Myc-79Q constructs using polyethylenimine (PEI) transfection reagent (Polysciences, Warrintong, USA). Seventy-two hours after transfection, cells were harvested in M-Per Mammalian Protein Extraction Reagent (Thermo Fisher Scientific, Waltham, USA) followed by lysis of adherent mammalian cells and procedure for IP of Myc-tagged proteins using Pierce Anti-Myc Agarose as described in the instruction manual (Thermo Fisher Scientific, Waltham, USA). Recombinant proteins were eluted using Pierce IgG Elution buffer (Thermo Fisher Scientific, Waltham, USA).
Protein Extraction
For the establishment of the TR-FRET, mouse tissues and cells were homogenized in three different lysis buffers: RIPA buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% IGEPAL), TES buffer (20 mM Tris, pH 7.5, 2 mM EDTA, 100 mM NaCl) supplemented with TNES buffer (50 mM Tris pH 7,5, 2 mM EDTA, 100 mM NaCl, 1% IGEPAL), or PBS buffer (DPBS (1×) Dulbecco’s Phosphate-Buffered Saline with 1% Triton™ X-100). All buffers were supplemented with cOmplete (EDTA-free) Protease Inhibitor (Roche, Mannheim, Germany). For each buffer condition, 10 mg tissue or cells were homogenized mechanistically in 100 μl of the respective buffer using the VDI 12 homogenisator (VWR, Darmstadt, Germany). Afterwards, homogenates were incubated on ice for 30 min vortexing every 10 min. Homogenates were stored at − 80 °C for later TR-FRET analyzes. Part of the homogenates were centrifuged at 13 200g for 30 min. Lysates were transferred to new collection tubes supplemented with 10% glycerol and stored at − 80 °C for later western blot analyses. The Bradford protein assay was used to determine the total protein concentration from homogenates and lysates [62].
SDS-PAGE and Western Blot
Twenty to 30 μg of total protein from cell culture or mouse tissue lysates were supplemented with 4 × LDS sample buffer (1 M Tris pH 8.5, 2 mm EDTA, 8% LDS, 40% glycerol, 0.075% CBB G, 0.025% phenol red) in a ratio 3:1 and 0.1 M dithiothreitol. Samples were heat denatured for 10 min at 70 °C and afterwards, electrophoretically separated [63] using 8 to 10% Bis-Tris gels with the electrophoresis MOPS buffer (50 mM MOPS, 50 mM Tris pH 7.7, 0.1% SDS, 1 mM EDTA) at 100 V, 250 mA for 2–2.5 h. Proteins were blotted [64] on nitrocellulose membranes (Amersham Protran Premium 0.2 μm, GE Healthcare) using transfer buffer (25 mM Bicine, 25 mM Bis-Tris pH 7.2, 1 mM EDTA, 15% methanol) at 80 V and 250 mA for 1.5 h. After transfer, the membranes were blocked with 5% skim milk powder in Tris-buffered saline (TBS) for 1 h, washed with TBS-T (TBS with 0.1% Tween 20) and incubated overnight at 4 °C with the following primary antibodies diluted in TBS-T: ataxin-2 polyclonal antibody (1:1000, 21776-1-AP, rabbit, Proteintech Group, Rosemont, USA); mouse anti-β-actin monoclonal antibody (1:500, clone AC-15, Sigma-Aldrich, Darmstadt, Germany). Afterwards, membranes were washed with TBS-T and incubated at RT for 1 h with secondary IRDye antibodies goat anti-mouse 800CW or goat anti-rabbit 800CW (both 1:5000, Li-Cor Biotechnology GmbH, Bad Homburg, Germany), respectively. After washing with TBS-T, fluorescence signals were detected using the LI-COR ODYSSEY FC and quantified with Image Studio 4.0 software (both Li-Cor Biotechnology GmbH, Bad Homburg, Germany). All full western blot images are provided as supplementary information figure 3a-f.
TR-FRET
To establish a polyQ-expanded ATXN2 specific time resolved fluorescence energy transfer (TR-FRET)-based immunoassay, two different ATXN2 antibodies and two different polyQ-specific antibodies were compared. Therefore, the two ATXN2 specific antibodies ataxin-2 polyclonal antibody (21776-1-AP, Proteintech Group, Rosemont, USA) and purified mouse anti-ataxin-2 monoclonal antibody (AB_398900, Becton, Dickinson and Company, Sparks, USA) were labeled with the fluorophore Tb by the company CisBio Inc. (PerkinElmer, Waltham, USA). Additionally, the two polyQ-specific antibodies clone MW1 (AB 528290, Development Studies Hybridoma Bank, Iowa, USA), and clone 5TF1-1C2 (MAB1574, Sigma-Aldrich, Darmstadt, Germany) received the acceptor fluorophore D2 by the same company. For establishment of the TR-FRET-based immunoassay, the labeled antibodies were diluted in detection buffer (50 mM NaH2PO4, 400 mM NaF, 0.1% BSA, 0.05% Tween-20) in different concentrations (Tb-labeled antibodies: 0.3 ng, 0.5 ng, 1 ng and D2-labeled antibodies: 1 ng, 3 ng, 10 ng). Mouse tissue or cell culture homogenates were diluted to a total protein concentration of either 2 μg/μl or 1 μg/μl in one of the following buffers (RIPA, TES/TNES or PBS) supplemented with cOmplete (EDTA-free) Protease Inhibitor. Human serum samples were measured undiluted or diluted 1:2 or 1:4 with PBS supplement with cOmplete (EDTA-free) Protease Inhibitor, respectively. Five microliters per diluted sample was incubated in duplicates with 1 μl antibody-mix (Tb-antibody with D2-antibody, ratio 1:1) in a low-volume white ProxiPlate 384 TC Plus plate (PerkinElmer, Waltham, USA) for 24 h at 4 °C. Signals were detected at 620 nm and 665 nm with the Multimode Plate Reader Envision (PerkinElmer, Waltham, USA), and the ratio between 665/620 was normalized to the total protein concentration and over the background signal (deltaF).
Statistical Analysis
The program GraphPad Prism 8 (GraphPad Software Inc., San Diego, USA) was used for the statistical evaluation of all data and for data visualization. Limit of detection (LOD) and quantification (LOQ) were calculated using GraphPad Prism software with a nonlinear regression model using a two-site binding saturation curve fit (including specific and background signal). The LOD is defined as the lowest concentration giving a signal greater than the background signal (above 3 standard deviations). The LOQ is defined as the lowest concentration giving a signal greater than the background signal (+ 10 standard deviations).
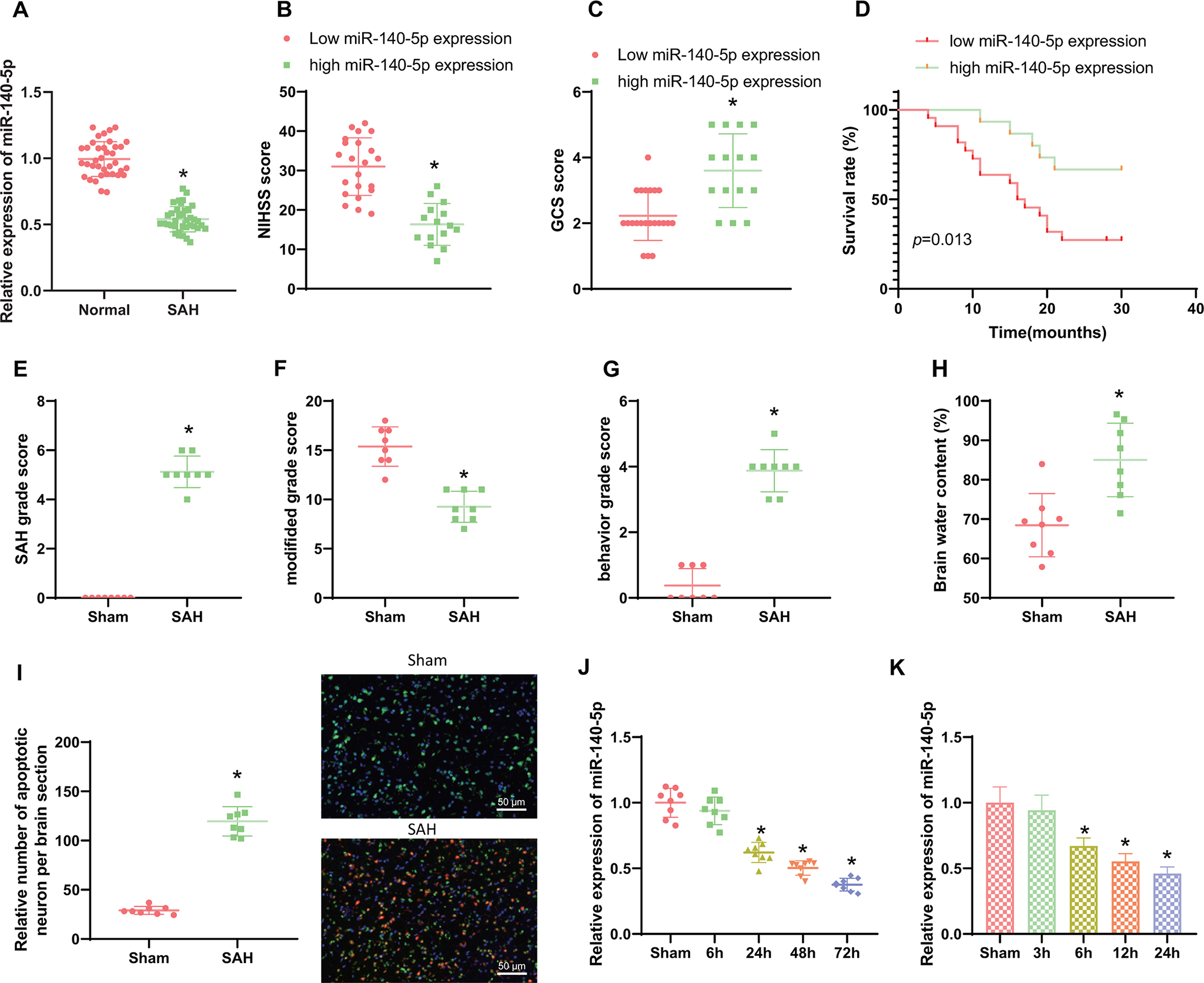
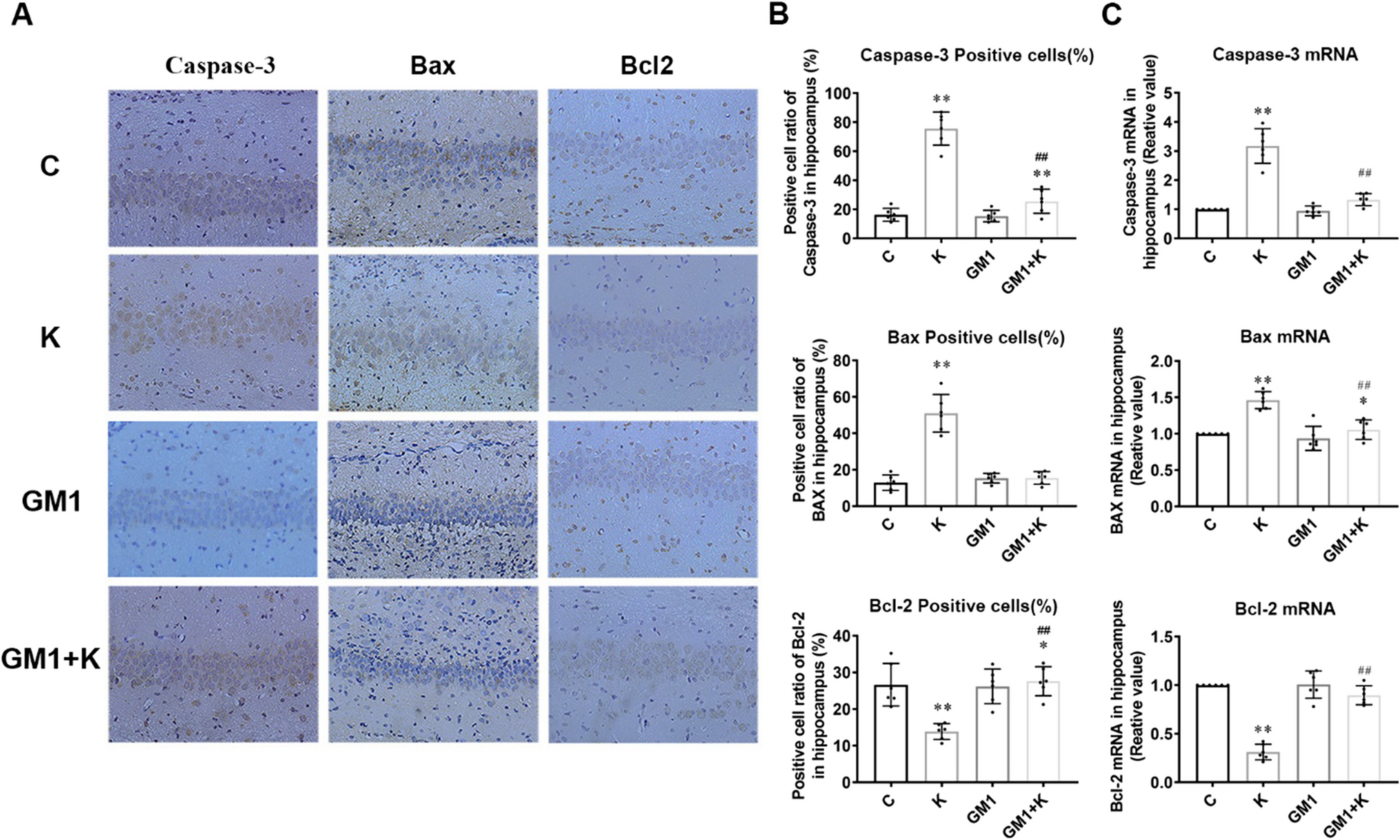
Normality of the datasets was evaluated by Shapiro-Wilk test. Due to the non-normally distributed data, the non-parametric Mann-Whitney U test was used to compare the different groups. P-values with less than 0.05 were considered statistically significant with *p < 0.05, **p < 0.01, and ***p < 0.001. All values are shown as mean ± standard error of the mean, SEM.





留言 (0)