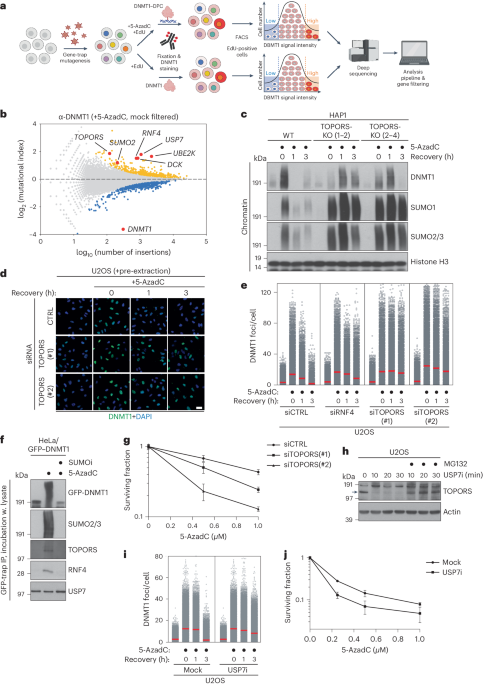
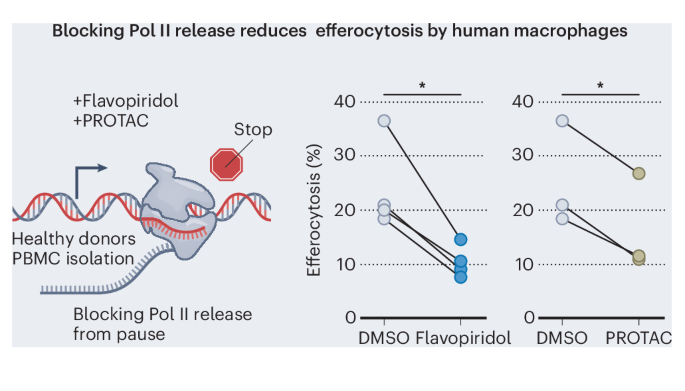
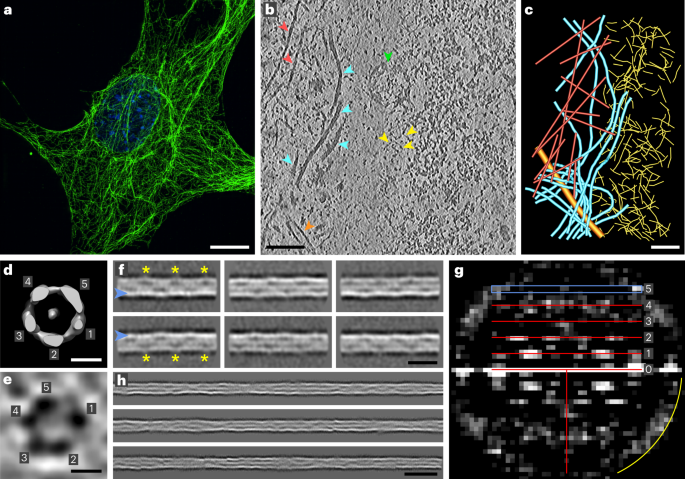
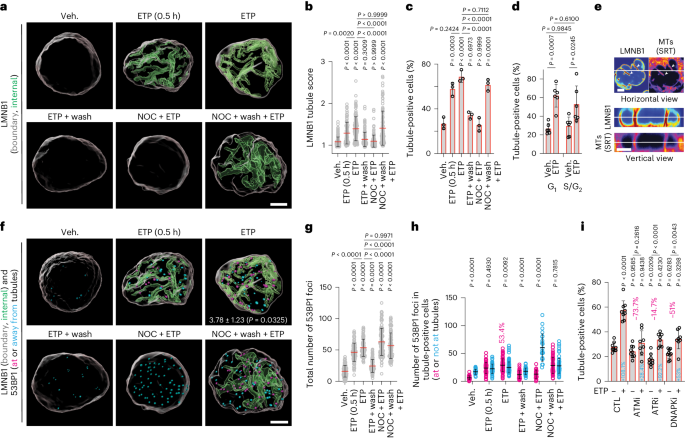
Genome editing and cell culture
HAP1 cells were cultured in Iscove’s modified Dulbecco’s medium (Invitrogen), containing 10% fetal bovine serum (Clontech), 1% UltraGlutamin (Lonza) and 1% penicillin–streptomycin (Invitrogen). Mutant cells were generated by CRISPR–Cas9 technology. Guide RNAs targeting exon 6 of SGO1 (primer, 5′-TGATGCTTACAATTTTAATT-3′), exon 10 of STAG1 (5′- TTGGCTGGACTCTTCATGAC-3′) and exon 11 of STAG2 (5′-GACAGTTATTTAAAATATGT-3′) were annealed into pX330. To mutate the locus of interest, we cotransfected a 100–120 base pair repair oligonucleotide with the desired mutation as well as a silent mutation: for SGO1Y335A F337A (5′-CAAAAAAAAATGCACAAATCTGTCAGTTCCAATGATGCTGCCAATGCTAATTTGGAAGAAGGTGTTCATCTTACTCCTTTCCGACAAAAAGTGAGCAATG-3′), STAG1W337A (5′-AGTACTGAGACAAACATAACTTCCATCAAAGCTTAGAACAG AGTAACTTACCCTGTCGTGAAGAGTAGCGCCAACATATTTTAGGTAACTGTCATTTAGGAAGGCATCACTATACATTTTCATC-3′) and STAG2W334A (5′-CTTAATGACAGTTATTTAAAATATGTTGGTGCGACTATGCATGATAAGGTAAGATGTGCCCTTCAGACTGCTTCTTTCTATACATCGGCGTGGCTGTCTGCACCTCTCATTCATGAG-3′). We cotransfected pBabePuro at a ratio of 1:10 to the pX330 plasmid. Cells were treated with 2 μg μl−1 puromycin for 2 days for selection. Colonies were picked, genomic DNA of clones was isolated and mutations were validated by Sanger sequencing.
siRNA transfection
All siRNAs were manufactured by Dharmacon (ON-TARGETplus). For SGO1 and luciferase we used SMARTpools, and for WAPL we used the following sequence: 5′-CAACAGUGAAUCGAGUAAUU-3′. Transfection was performed with 20 μM per siRNA final concentration, using Invitrogen RNAiMAX (Life Technologies), following the manufacturer’s instructions.
Chromosome spreads
Cells were transfected with the corresponding siRNAs, and after 2 days the cells were treated with nocodazole as described previously31. Images were randomized by a homemade ImageJ macro and then visually assigned their corresponding phenotype. A parametric two-tailed t test was used to compare the scoring of cohesion phenotypes.
Immunofluorescence
For immunofluorescence, cells were treated with nocodazole, fixed and stained as described previously31. For immunofluorescence spreads, cells were treated with the corresponding siRNA. After 24 h, cells were transfected using FuGENE transfection reagent (Promega) with 0.8 μg SGO1–GFP plasmid (kindly provided by S. Lens) or a SGO1Y335A F337A–GFP mutant plasmid. One day later, cells were treated with nocodazole for 1.5 h or with MG132 for 2 h, and mitotic cells were collected by shake-off. Cells were washed once in phosphate-buffered saline, followed by a quick spin onto microscope slides with a Shandon Cytospin centrifuge. Cells were extracted with PBS containing 0.3% Triton-X for 5 min and fixed in 4% paraformaldehyde for 15 min. The coverslips were washed three times with PBS containing 0.1% Triton-X before being incubated with antibodies at a 1:1000 dilution in PBS containing 3% BSA and 0.1% Triton-X overnight at 4 °C. Secondary antibody incubations were performed by incubation at room temperature for 1 h with DAPI in PBS containing 3% BSA and 0.1% Triton-X. Coverslips were mounted in Prolong Gold (Invitrogen).
Images were obtained using a DeltaVision deconvolution microscope (Applied Precision), and images were acquired using Softworx (Applied Precision) and ImageJ. To establish levels of SGO1 in prometaphase cells, we used an ImageJ macro that allowed us to calculate the level of SGO1 relative to CENPA. To identify the location of SGO1–GFP in mitotic cells, we first blinded the channel corresponding to GFP to prevent bias towards a phenotype. Next, we drew a straight line on four random chromosomes that showed two distinct centromeres and obtained the plot profile of both CENPA and GFP for each location.
Live-cell imaging
Cells were grown on glass-bottomed dishes (LabTek). To visualize the DNA, 2 h before imaging, a SiR-DNA probe (1:2000, Spirochrome) was added. Images were taken using a DeltaVision deconvolution microscope (Applied Precision). Cells were imaged every 5 min using a ×40 air objective with 4 × 2.5 μm Z stacks. Images were acquired using Softworx (Applied Precision) and ImageJ.
Fluorescence in situ hybridization
Prometaphase samples cells were obtained as described above. Fixed cells were dropped on cover slides and then dried. We added probes against the centromere of chromosome 8 (XCE 8 ORANGE, MetaSystems Probes) and shielded the cells with a coverslip and rubber cement. The slides were incubated for 2 min at 75 °C, followed by overnight incubation at 37 °C. The cells were washed with 0.4× SSC at 72 °C for 2 min, followed by washing at room temperature with 2× SSC, 0.05% Tween-20, for 30 s. The slides were washed with water and stained with DAPI, followed by mounting with Prolong Gold (Invitrogen).
G2 samples were collected by treating the cells for 18 h with RO-3306. We verified that the cells were synchronized in G2 by incubation in Nicoletti buffer followed by flow cytometry (BD LSRFortessa). Plots were generated with FlowJo (v.10). G2-synchronized cells were spun down and resuspended with fixative solution (methanol/acetic acid, 3:1), followed by the same protocol as described above.
Images were taken using a DeltaVision deconvolution microscope (Applied Precision), and images were acquired using Softworx (Applied Precision) and ImageJ. The fluorescence signal was categorized as singlet (distance between the two highest intensity signals ≤300 nm) or doublet (distance between the two highest intensity signals >300 nm), as described previously32.
Immunoblotting and coimmunoprecipitation
Immunoblot and coimmunoprecipitation were performed as previously described33.
Antibodies
The following antibodies were used as primary antibodies for immunofluorescence microscopy: SGO1 (SAB1405371, Sigma Aldrich), GFP (ab290, Abcam) and CENPA (07–574, Millipore; and ab13939, Abcam). For immunoblotting, the following primary antibodies were used: SA1 (ab4457, Abcam), SA2 (A300-158a, Bethyl Laboratories), SMC1 (A300-055A, Bethyl Laboratories), SCC1 (05-908, Millipore), WAPL (A-7, sc-365189, Santa Cruz), Sororin (ab192237, Abcam), HSP90 (sc-13119(F-8), Santa Cruz) and α-tubulin (T5168, Sigma Aldrich). All primary antibodies were used at a 1:1000 dilution with the exception of HSP90 and α-tubulin (1:10000). For coimmunoprecipitation, we used 4.5 μg of SMC1 (A300-055A, Bethyl Laboratories) or IgG (2729 S, Cell Signaling) per sample. Secondary antibodies were used at a 1:1000 dilution. For immunofluorescence microscopy we used: Alexa Fluor 488 goat anti-mouse (A-11001, Life Technology), Alexa Fluor 568 goat anti-mouse (A-11004, Life Technology), Alexa Fluor 488 goat anti-rabbit (A-11008, Life Technology) and Alexa Fluor 568 goat anti-rabbit (A-11011, Life Technology). For western blots, we used the following secondary antibodies: anti-goat-PO (P0449, DAKO), anti-rabbit-PO (P0448, DAKO) and anti-mouse-PO (P0447, DAKO).
Constructs, protein expression and purification
SA2 amino acid residues 80–1060 were expressed as a GST fusion protein and SCC1 amino acid residues 281–420 as an N-terminally 6×His-tagged fragment as described previously21. Expression and purification were done as described previously21. SGO1 constructs were cloned into the BamHI and NotI sites of pGEX-6P1. Mutagenesis was done using a Q5 Site-Directed Mutagenesis Kit (New England Biolabs). All proteins were expressed in Escherichia coli BL21 (DE3) by autoinduction, and purification was done as described previously21.
Crystallization and structure determination
Crystallization of the SA2–SCC1 complex was done as described previously18,21. Crystals were soaked for 7 days with a 500 μM peptide solution including SGO1 amino acid residues 331–341 SNDAYNFNLEE. Crystals were cryoprotected as described previously21. Diffraction data were collected at 100 K at an X-ray wavelength of 0.9687 Å at beamline ID30A-1/MASSIF-1 (ref. 34) of the European Synchrotron Radiation Facility, with a Pilatus3 2M detector, using automatic protocols for the location and optimal centering of crystals35.
Data were processed with XDS36 and imported into CCP4 format using AIMLESS37. The structure was determined by molecular replacement using Phaser (Phenix 1.14-3260)38. A final model was produced by iterative rounds of manual model-building in Coot (COOT 0.8.0-3)39 and refinement using PHENIX (1.14-3260)40. The SA2–SCC1–SGO1 model was refined to a resolution of 3.2 Å with Rwork and Rfree values of 25% and 28%, respectively (Table 1). Structures were rendered with PyMOL (2.2.3). Analysis with MolProbity (4.3)41 showed that there were no residues in disallowed regions of the Ramachandran plot, and the all-atom clash score was 12.3 (63rd percentile). The computational model shown in Fig. 1c was calculated using AlphaFold v.2.1.1 with multimer model v1 weights42. The computational model shown in Fig. 2b was generated by superposition of an AlphaFold model for SA1 onto SA2 in the SA2–SCC1–SGO1 complex.
GST pulldowns
GST pulldowns were done as described previously21 with small modifications. Briefly, 50 μM GST-tagged SGO1 constructs were mixed in 50 μl buffer 1 (20 mM Tris-HCl, pH 7.8, 500 mM NaCl, 0.5 mM TCEP, 0.1% Tween-20) containing 25 μl of a 50% slurry of GST Sepharose beads (Cytiva) per reaction. GST beads were incubated for 1 h at 4 °C, followed by four washes with 500 μl of buffer 1. Then, 2.5 μM of SA2–SCC1 was added, followed by overnight incubation at 4 °C. A 25-μl volume of the reaction was withdrawn as the reaction input, and the remainder was washed five times with 500 μl of buffer 1. Samples were boiled in 1× sodium dodecyl sulfate (SDS) sample loading buffer (New England Biolabs) for 5 min to obtain the bound fraction, followed by SDS polyacrylamide gel electrophoresis analysis. ITC was performed as described previously21. ITC data were analyzed with Origin 7.0.
Statistics and reproducibility
No statistical method was used to predetermine the sample size. No data were excluded from the analyses. All experiments with phenotype calling were randomized, and the SGO1 signal was blinded in all experiments for SGO1 localization with respect to the centromere. Data were visualized with Prism 9. For all pairwise comparisons, we performed t test analyses, with a probability threshold of P = 0.05. GST pulldowns were repeated at least three times with consistency.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.





留言 (0)