
記住我

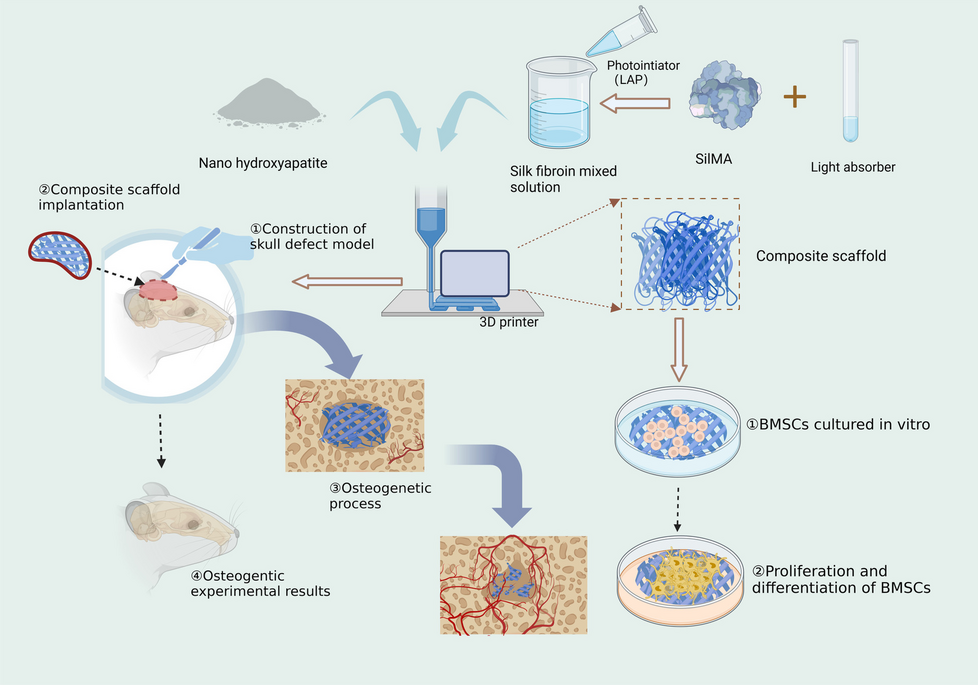
It is possible to use virtual screening to discover the best intermolecular framework between macromolecular targets and small molecules, such as drugs via the screening of chemical compound databases. It predicts which ligand will interact optimally with a target to form a complex. The ligand is then sorted according to its binding free energy with the target. The present study demonstrated the utilization of ZINC database natural compounds (n = 97,999) to conduct a virtual screening against the PEDV 3CL protease to identify putative hits. The binding free energy of each compound included in the study was evaluated to identify putative hits for further evaluation. Generally, a protein-ligand complex with low binding energy has a high binding affinity. Therefore, the top 10 screened compounds exhibiting minimum binding energy with the 3CL protease were selected as putative lead compounds (-11.0 to -9.9 Kcal/mol). The binding energy of the co-crystallized ligand GC376 was − 7.5 kcal/mol, and the RMSD value between GC376 and its re-docked conformation was predicted to be 1.224 Å. This RMSD value serves as an indicator of the efficiency and validity of the docking protocol. The ZINC id, binding free energy, and interacting amino acid that contributed to the protein-ligand interactions of top-10 lead like compounds are listed in Table 1.
Table 1 List of the top 10 screened compounds, their binding free energies, and interacting amino acid residues of the PEDV 3CL protease. The amino acid residues in bold indicate the residues involved in conventional hydrogen bond interactions Evaluation and visualization for the binding of lead compounds with 3CL proteaseVirtual screening offers the best interacting compounds with macromolecular targets based on binding free energy. Of the top 10 screened compounds, the top five were selected (binding energy range: -11.0 to -10.1 kcal/mol) for further analysis using different parameters to evaluate its binding nature and inhibitory potential with the 3CL protease. ZINC38167083 was found to interact with Arg130 and Leu283 with a conventional hydrogen bond; forming one pi-sigma and one alkyl bond with Leu198; one pi-pi t-shaped and one pi-alkyl with Tyr280, one carbon-hydrogen bond with Ser282, and two pi-anion bonds with Asp285 with 3CL protease and a binding free energy of -11.0Kcal/mol (Fig. 2A). Moreover, ZINC09517223 interacted with the 3CL protease and Asn141, which has one conventional hydrogen bond; His41 with two pi-pi t-shaped bonds; Thr47 with one pi-sigma bond and Leu164 with two pi-alkyl bonds and a binding free energy of -10.4 Kcal/mol (Fig. 2B). ZINC03960761 interacted with 3CL protease with a binding energy of -10.3 Kcal/mol and formed one conventional hydrogen bond with Gln187; one carbon-hydrogen bond with Gln163 and Thr189; one pi-pi t-shaped bond with His41 and one pi-sigma bond with Thr47, as well as two pi-alkyl bonds with Leu164 (Fig. 2C). ZINC04339983 interacted with 3CL protease with a binding energy of -10.2 Kcal/mol and formed one conventional hydrogen bond with Asn141, and one carbon-hydrogen bond with Glu165; two pi-pi t-shaped bonds with His41; one pi-sigma bond with Thr47; one amide-pi stacked bond with Ile140; one pi-alkyl bond with Ala143; one pi-alkyl and pi-sulfur bonds with Cys144, as well as two pi-alkyl bonds with Leu164 (Fig. 2D). ZINC09517238 interacted with the 3CL protease with a binding energy of -10.1 Kcal/mol. It formed one conventional and one carbon-hydrogen bond with Asn141 and Glu165, respectively. It also interacted with His41 with two pi-pi t-shaped bonds; Thr47 with one pi-sigma bond; Ile140 with one amide-pi stacked bond; Ala143 with one pi-alkyl bond; Cys144 with one pi-alkyl and one pi-sulfur bond; Leu164 with two pi-alkyl bonds; Besides, it formed one alkyl bond with Leu190 (Fig. 2E). Whereas, the interaction of reference compound i.e. co-crystallized ligand GC376 was also analyzed. It was interacted with 3CL protease Phe139, His162, and Glu165 with conventional hydrogen bonds; His41 with alkyl bond; Leu164 with alky and pi-alkyl bonds as well as Leu166 with pi-alkyl bond but binding energy was observed as higher (-7.5 Kcal/mol) as compared to identified natural putative lead compounds.
Fig. 2
2D representation of the binding interactions of the top five screened natural compounds with PEDV 3CL protease depicted key amino acid residues that contributed to protein-ligand interactions. (A) ZINC38167083, (B) ZINC09517223, (C) ZINC03960761, (D) ZINC04339983, (E) ZINC09517238
Assessment of drug-likeness through the physicochemical property, toxicity, and PAINS analysisThe drug-likenesses of the top five selected compounds were analyzed based on ADMET analysis followed by PAINS. In any drug discovery program, ADMET analysis represents a fundamental task to investigate the chemical nature of the compounds. Therefore, different parameters such as molecular weight, LogP, H-bond donor and acceptor, topological polar surface area, mutagenic, tumorigenic, and irritant properties as well as the PAINS of each selected compound were analyzed. ADME and PAINS-related information were extracted from the ZINC database. In addition, the OSIRIS Property Explorer tool was used to predict toxicity (T). Based on our analysis, the top-screened selected compounds exhibited drug-like properties but PAINS-related problem in ZINC03960761 and irritation in ZINC09517238 was detected during PAINS and toxicity analysis, respectively. Besides, all the selected compounds also possessed polar surface areas < 140 Å2, indicating high cell membrane permeability. The results obtained during the drug-like analysis are depicted in Table 2.
Table 2 Physicochemical properties and drug-likeness from the identified compounds Structural and conformational analysis of the 3CL protease in unbound and bound systems during MD simulationTo visualize the dynamic behavior of the 3CL protease both before (unbound) and after ligand binding (bound), a 500 ns MD simulation was conducted. Various parameters i.e., RMSD, RMSF, Rg, SASA, H-bond, PCA, FEL, and the binding free energy calculation were used to summarize the results.
Stability analysisRMSD was used to measure conformational stability during the MD simulation of proteins in relation to their structure. Specifically, structures with smaller RMSD values are more stable than those with larger RMSD values. We have computed the RMSD value for 500 ns (Fig. 3A). Deviation from initial to next and subsequent structures is represented by it. Here, based on the RMSD graph of backbone c-alpha atoms, the 3CL protease, and all the complexes exhibited fewer fluctuations with low RMSD values following 400 ns. The average RMSD of the 3CL protease was calculated as 0.26 nm. Moreover, the RMSD values of the 3CL protease-ZINC38167083, 3CL protease-ZINC09517223, 3CL protease-ZINC03960761, 3CL protease-ZINC04339983, and 3CL protease-ZINC09517238 were 0.49, 0.41, 0.47, 0.29, and 0.26 nm, respectively. It was predicted that all complexes got established after 400 ns. Hence, the final 100 ns trajectories were considered for further analysis.
Flexibility analysisProteins maintain their properties by being flexible, which can be accessed through RMSF analysis. The RMSF of the 3CL protease and their complexes were therefore analyzed for the last 100 ns equilibrated trajectory. This indicates amino acid residue fluctuations upon binding of ligands, here higher fluctuations were observed for amino acid residues from positions 180 to 299 than for amino acid residues from positions 1 to 179. Although the mean RMSF value for most amino acid residues was below 0.4 nm. Besides, Ala1, Gly298, and Val299 exhibited higher RMSF mean values (Fig. 3B). The average RMSF value of the 3CL protease was measured as 0.12 nm. Moreover, the RMSF values of the 3CL protease-ZINC38167083, 3CL protease-ZINC09517223, 3CL protease-ZINC03960761, 3CL protease-ZINC04339983, and 3CL protease-ZINC09517238 were 0.16, 0.15, 0.21, 0.14 and 0.13 nm, respectively (Fig. 3B). Overall, lower fluctuation was observed except for the 3CL protease-ZINC03960761 complex, supporting their utilization as the putative lead compounds.
Compactness analysisTo understand protein structure compactness, stability, and folding, the Rg values can be calculated over time. The 3CL protease and their complexes were analyzed for Rg values to determine their structural compactness. The Rg values of the 3CL proteases were calculated and plotted, the 3CL protease-ZINC38167083, 3CL protease-ZINC09517223, 3CL protease-ZINC03960761, 3CL protease-ZINC04339983 and 3CL protease-ZINC09517238 systems from the final 100 ns MD trajectories. The average Rg values were calculated as 2.14, 2.15, 2.20, 2.13, 2.19, and 2.15 nm, respectively. The Rg results indicate that the 3CL protease-ZINC03960761 exhibits a more compact structure than the other complexes (Fig. 3C).
Solvent accessible surface area (SASA) analysisLigand-induced solvent-accessible area changes were determined using a SASA analysis from the final 100 ns of the simulation (Fig. 3D). The average SASA value for the 3CL proteases, 3CL protease-ZINC38167083, 3CL protease- ZINC09517223, 3CL protease–ZINC03960761, 3CL protease-ZINC04339983, and 3CL protease-ZINC09517238 was calculated as 140, 146, 148, 146, 151, and 146 nm2, respectively. Here, the SASA value of the 3CL protease-ZINC04339983 complex was higher than the others. Besides, a similar type of pattern was observed for all the systems (Fig. 3D), revealing comparatively least changes following the binding of each compound.
Fig. 3
Stability analysis (A) RMSD values for the PEDV 3CL protease-compound complexes. Flexibility analysis (B) RMSF values for the PEDV 3CL protease-compound complexes over the final 100 ns of the simulations. Compactness (C) Rg, and Solvent accessible surface area analysis (D) SASA values for the final 100 ns of the simulations. Black, red, green, blue, yellow, and maroon colors represent the PEDV 3CL proteases, ZINC38167083, ZINC09517223, ZINC03960761, ZINC04339983, and ZINC09517238, respectively
Hydrogen bonds (HBs) analysisThe most crucial bond that stabilizes the protein-ligand interactions is the hydrogen bond. Therefore, we counted the number of hydrogen bonds produced from the natural compounds’ interaction with the target 3CL protease. Here, the 3CL protease-ZINC38167083 exhibited the highest number (0–8) of hydrogen bonds compared to the estimated complexes (Fig. 4).Comparatively, the 3CL protease-ZINC09517223 complex showed an average of 0–5 hydrogen bonds; the 3CL protease-ZINC03960761 complex averaged 0–2 hydrogen bonds; 3CL protease-ZINC04339983 averaged 0–4 hydrogen bonds, and the 3CL protease-ZINC09517238 averaged 0–5 hydrogen bonds during the final 100 ns (Fig. 4). Thus, during the protein-ligand interactions, these compounds interacted with the 3CL protease to produce a stable complex.
Fig. 4
Changes in the number of hydrogen bonds in each respective complex according to data from the final 100 ns of the simulations. Red, green, blue, yellow, and maroon colors represent ZINC38167083, ZINC09517223, ZINC03960761, ZINC04339983, and ZINC09517238, respectively
Principal component analysis (PCA)PCA analyses were conducted to capture significant conformational changes during ligand binding. A protein’s overall motion is determined primarily by the first few eigenvectors. Therefore, changes in the structural movement were analyzed using the first 50 eigenvectors (Fig. 5A). The percentage-wise correlated motions were calculated from the initial 10 eigenvectors and provided a clear understanding of the motions induced by the ligand binding. The 3CL proteases, 3CL protease-ZINC38167083, 3CL protease- ZINC09517223, 3CL protease-ZINC03960761, 3CL protease-ZINC04339983 and 3CL protease-ZINC09517238 showed 69.57%, 76.18%, 64.51%, 84.11%, 62.45% and 68.17% correlated motions, respectively. Here, we can see that the 3CL protease-ZINC09517223 and 3CL protease-ZINC04339983 complexes showed the lowest motions.
Fig. 5
Principal component analysis. (A) Eigenvalues derived from the final 100 ns of each simulation and used for PCA depicted eigenvalues vs. the first fifty eigenvectors. (B) First two eigenvectors depicted the PEDV 3CL protease motion in space for all the systems. Black, red, green, blue, yellow, and maroon colors represent PEDV 3CL protease, ZINC38167083, ZINC09517223, ZINC03960761, ZINC04339983, and ZINC09517238, respectively
Figure 5 illustrates that the first few eigenvectors reflect the overall dynamics of the protein. This led to the selection of the first two eigenvectors and their plotting in phase space (Fig. 5B). The 3CL protease, 3CL protease-ZINC38167083, 3CL protease- ZINC09517223, 3CL protease-ZINC04339983, and 3CL protease-ZINC09517238 clusters are the most stable (low correlated motions) when compared to the 3CL protease–ZINC03960761(Fig. 5B).
Gibbs free energy landscape (FEL) analysisGibbs free energy landscape calculations were performed using the first two principal components. Figure 6 illustrates the FEL calculated for all the systems. The color bar represents the Gibbs free energies (kcal/mol) ranging from the lowest energy (blue) to the highest energy (dark yellow) conformational states. In the case of 3CL proteases, 3CL protease-ZINC38167083, 3CL protease-ZINC09517223, 3CL protease-ZINC04339983, and 3CL protease-ZINC09517238, the enriched energy minima with wide space have been observed (blue color). These cover a larger blue area as compared to the 3CL protease–ZINC03960761 complex and represent a stable cluster (Fig. 6). Moreover, there are several minima in the conformational space of 3CL protease-ZINC03960761 (Fig. 6D). Additionally, FEL analysis demonstrated that all the complexes gained minimum energy corresponding to the most stable conformations.
Fig. 6
The color-coded illustration of the Gibbs free energy landscape (FEL) was plotted using PC1 and PC2. The color bar indicates the Gibbs free energies (kcal/mol) for conformational states with the lowest (blue) and highest (red) energies. (A) PEDV 3CL protease (B) 3CL protease-ZINC38167083, (C) 3CL protease-ZINC09517223, (D) 3CL protease-ZINC03960761, (E) 3CL protease-ZINC04339983, and (F) 3CL protease-ZINC09517238 docked complexes
Binding free energy calculationAn MM-PBSA method was used to estimate the binding free energy of all simulated complexes to validate their binding affinities. Binding free energies were calculated using the last 5 ns of the MD simulation trajectories. The calculated binding free energy for the 3CL protease-ZINC38167083, 3CL protease-ZINC09517223, 3CL protease-ZINC03960761, 3CL protease-ZINC04339983, and 3CL protease-ZINC09517238 complexes were − 104.208, -133.292, -176.620, -168.155 and − 153.980 kJ mol− 1, respectively. The calculated values for the van der Waals, electrostatic, polar solvation, SASA, and binding free energies are given in Table 3.
Residual binding energy analyses were conducted for the simulated complexes to identify the amino acid residues that are crucial for ligand binding. All the selected compounds were observed to be significantly involved in the protein-ligand interactions with amino acid residues of the 3CL protease, which indicates a potential for 3CL protease inhibitors. The amino acid residues from positions 1 to 50 and 135 to 195 contributed more to interactions (Fig. 7).
Table 3 Affinities of natural compounds with a PEDV 3CL pro (van der Waals and electrostatic forces, polar solvation, SASA, and binding free energy in kJ mol-1) Fig. 7
Plot depicting the amino acid residues in the PEDV 3CL proteases contributing to the binding with natural compounds. Red, green, blue, yellow, and maroon colors represent ZINC38167083, ZINC09517223, ZINC03960761, ZINC04339983, and ZINC09517238, respectively
留言 (0)