Transgenic mice
Mice were housed under specific standardized pathogen-free conditions in the animal facility at Ulm University. Food and water were provided ad libidum, a 12light/12dark cycle was obeyed. IKK2-CAPLP−CreERT2 was generated by crossing single transgenic Tg(Plp1-cre/ERT2)1Ueli [82] mice (short PLP-CreERT2) with mice single transgenic for Gt(ROSA)26Sortm4(Ikbkb)Rsky [83]. The RFPPLP−CreERT2 Cre reporter line was established by crossing single transgenic Tg(Plp1-cre/ERT2)1Ueli [82] mice with mice single transgenic for Gt(ROSA)26Sortm1Hjf [84]. The RFPPLP−CreERT2 model allows a direct in situ monitoring of recombination-dependent RFP expression i.e., cellular deletion efficiency. All mouse lines were bred on a C57BL/6 background. Activation of the genetic construct was conducted at the age of 9–13 weeks by 3 intraperitoneal (i.p.). injections of tamoxifen (2 mg tamoxifen dissolved in sun flower seed oil, Sigma-Aldrich, St. Louis, MO, USA) within 5 days and 14 days of tamoxifen enriched food (400 mg tamoxifen citrate/ 1 kg, Genobios, Laval, France), Fig. S1b. Thereafter animals were observed up until 40 weeks post induction (wpi). Wild-type and single transgenic littermates were used as control and just like the double transgenic animals subjected to tamoxifen administration, both male and female animals were included in the study.
All animal experiments were performed in compliance with the guidelines of the German Animal Protection Act and were approved by the Regierungspräsidium Tübingen.
Behavioural analysesNeurological Severity Score (NSS)
A 10-point NSS originally established to monitor neurological deficits after traumatic brain injury (TBI) was used to assess neurological deficits [34]. This scoring system consists of 10 tests, including tasks to measure cognitive and motor functions (e.g., beam walk, round-stick balance, exit circle, gait pattern, and exploratory interest in new environment), whereby 1 point is given for failure of the task and 0 points for succeeding. Thus, a maximum NSS of 10 points indicates severe neurological dysfunction, with failure of all tasks. In the present study, the NSS was assessed starting post induction at least every 4 weeks up to 40 wpi.
Rotarod
Motor behavior was analyzed with the ENV-575 M rotarod (Med Associates, St. Albans, VT, USA). After 1 min at 4 rpm for adjustment, the cylinder accelerated within 5 min to 40 rpm. The latency until falling off the accelerating rotarod was recorded. For the trained group, pretraining was assessed in the second week of tamoxifen food (consisting of 3 trials each on 3 consecutive days) and analysis was performed at least every 4 weeks up to 40 wpi. One group was left untreated and firstly admitted to pretraining after 20wpi.
Beam walking test
In this test, the mice had to traverse a narrow beam to escape from a small, elevated platform to a closed dark box, with subtle encouragement by the experimenter. For the trained group, a protocol with 4 training trials per day for 3 consecutive days with a 12 mm square beam (length 100 cm) was used and analysis was done in duplicates on different beam sizes (12 mm square, 5 mm square, 17 mm round, 11 mm round) at least every 4 weeks up to 40 wpi. The untrained group was only admitted to 1 day of trials on the 12 mm square beam 20wpi and analyzed on the different beam sizes thereafter.
Grip strength measurement
Grip strength was measured using a grip strength meter at maximum strength mode (Panlab, Harvard Apparatus); 3 trials were recorded and averaged. Grip strength was recorded at least every other week.
Ladderwalk
The ladder consists of two transparent plates (square: 69.5 × 15 cm) and cylindrical ladder rungs (8 cm long, Ø of 2 mm). The ladder walk has a length of 60 cm with a space of 1 cm between rungs for the regular ladderwalk and spaces varying between 0.5 and 2 cm for the irregular ladderwalk. Each mouse crossed the beam four times consecutively, while recorded with a Samsung NX1000 camera. The recorded videos were analyzed at ¼ of the original speed to count slips.
Primary cell isolation
Primary OLs and microglia from adult mice were isolated using the Adult Brain Dissociation Kit, OctoMACS® with heaters, O4 and Cd11b beads as well as MS columns in a magnetic field (Miltenyi Biotec, Bergisch-Gladbach, Germany) according to the manufacturer’s protocol. In brief, brains were dissected, enzymatically digested, and debris was removed. The obtained cell suspension was labeled with O4 beads, washed, and then added to an MS column. The pellet of purified O4 positive OLs was snap-frozen in liquid nitrogen (N2) and stored at -80 °C until further analysis. The flowthrough and three washing volumes of the column containing unlabeled cells were combined and labeled again, this time with CD11b beads, washed and purified using an MS column. The pellet of purified CD11b positive microglia was snap-frozen in liquid nitrogen (N2) and stored at -80 °C until further analysis.
X-Gal staining
Frehsly isolated primary O4 + OLs were seeded as a droplet of 300μL with a concentration of 1 × 106 cells/mL in 0.5%BSA in PBS in the middle of a well in a 6-well plate. After cells were allowed to attach for 1.5 h at room temperature, the well was filled with 0.5%BSA in PBS very carefully before centrifuged at 200 g for 3 min at room temperature. Cells were then stained using the Senescence β-Galactosidase Staining Kit (Cell signaling #9860S) according to manufacturer´s instructions. In brief, cells were fixed for 10 min at room temperature and subsequently covered with staining solution and incubated at 37 °C for 24-48 h. If clear blue color formation was visible, cells were mounted with 70% glycerol. Images were taken with a Leica DM IRB Microscope (Wetzlar, Germany) and analyzed.
Protein extraction and immunoblot analysis
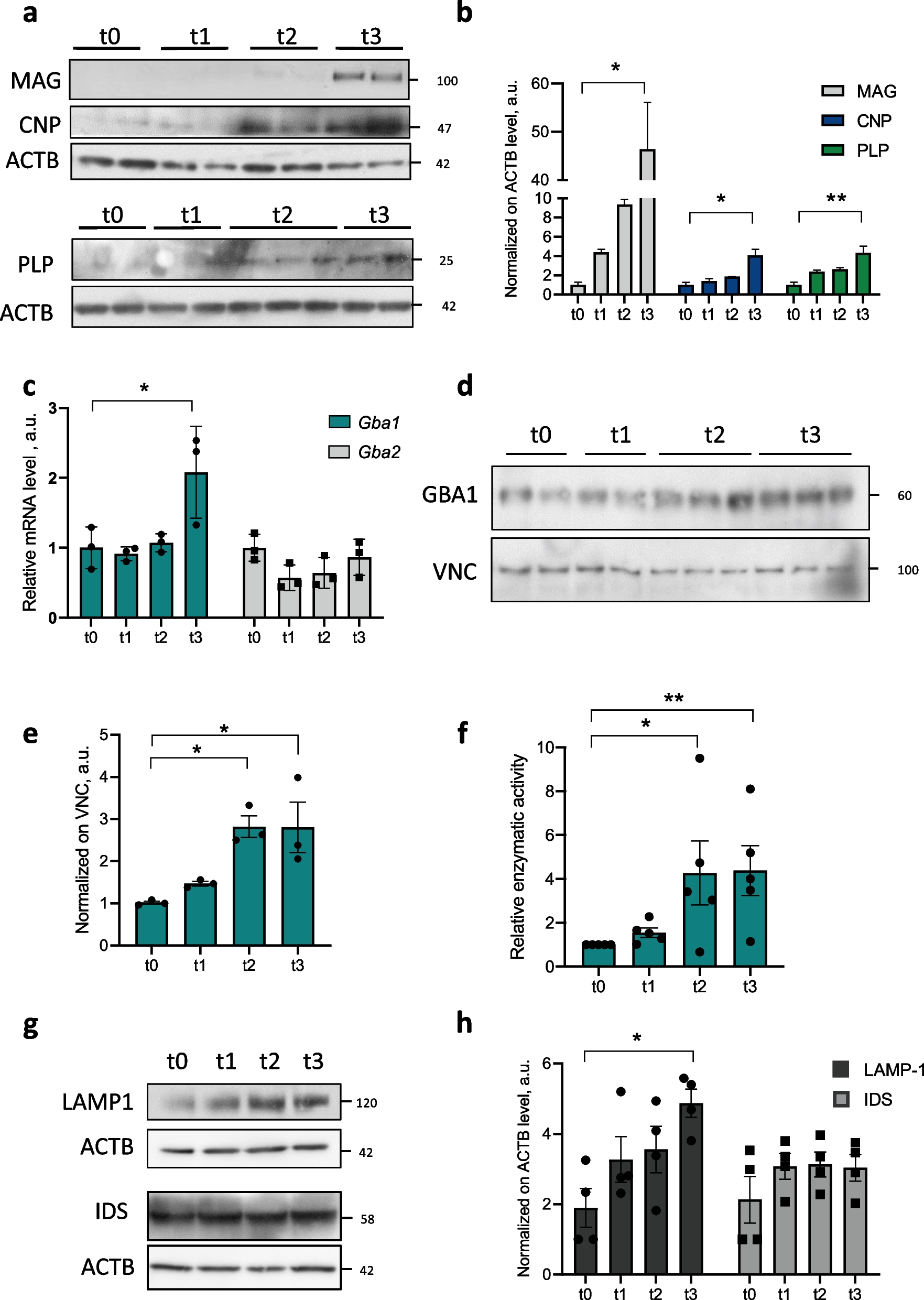
Tissue samples from different brain regions were snap-frozen in liquid nitrogen, pulverized with a pestle, aliquoted, and stored at -80 °C. For protein extraction an aliquot of one spatula was lysed in 200 μL (approximately three times its volume) of KA-lysis buffer (25 mM Tris–HCl, 150 mM NaCl, 25 mM sodium pyrophosphate, 50 mM β-glycerophosphate, 50 mM NaF, 2 mM EGTA, 2 mM EDTA, 1 mM DTT, 10% glycerol, 1% Triton X-100 (pH 8.0)) supplemented with protease inhibitors (1 mM PMSF and CompleteMini Tablet; Roche Diagnostics, Mannheim, Germany). After centrifugation (30 min, 13,000 rpm), the supernatant was used as the total protein extract. Samples were denatured with a fourfold concentrated Laemmli buffer (200 mM Tris–HCl, 15% glycerol, 4% SDS, 5% β-mercaptoethanol, bromphenol blue). Cell pellets were lysed at a concentration of 107 cells per 100 μL SDS sample buffer (62.5 mM Tris–HCl (pH 6.8), 2% SDS, 10% Glycerin, 50 mM DTT, 0.01% bromphenol blue) and sonicated 3 times for 5 s. Protein extracts from tissue samples as well as cell pellets were boiled for 5 min at 100 °C. Equal amounts of protein (50 μg of tissue samples and 25 μL of cell pellet samples) were separated by SDS-PAGE and transferred to nitrocellulose membranes via wet transfer. After blocking with 5% nonfat dry milk in TBS-T (0.05% Tween 20) buffer for 1 h at room temperature, primary antibodies (see below) were incubated in blocking solution overnight at 4 °C or for 2 h at room temperature. After 3 washing steps, membranes were incubated with horseradish peroxidase–coupled secondary antibody for 1 h at room temperature. Membranes were exposed to ECL detection reagent (Thermo Fisher Scientific, Waltham, MA, USA) and developed by ECL.
Histology and immunostaining
Two different techniques were used: a) Brains were fixed by immersion with 4% paraformaldehyde (PFA) overnight at 4 °C, dehydrated, embedded in paraffin, and cut to 7 mm–thick coronal sections on a microtome (Microm HM355S; Thermo Fisher Scientific). For immunofluorescence, after rehydration, heat mediated antigen retrieval was performed with sodium citrate (10 mM, pH 6.0, 0.05% Tween 20) and the tissue sections were additionally incubated with 0.5% Triton X-100 for 30 min. Sections were washed with PBS and blocked with 5% bovine serum albumin (BSA) for 1 h at room temperature. Incubation with primary antibodies (in 5% BSA) was performed overnight at 4 °C and incubated afterward with secondary antibodies (in 5% BSA) for 1 h at room temperature with 100 ng/ml DAPI for nuclear counterstaining. For histology using Luxol Fast Blue (LFB) and Nissl staining, after dehydration sections were incubated in 0.1% LFB solution at 60 °C overnight, rinsed in 95% ethanol and distilled water before incubated in 0.05% Lithium carbonate solution for 5 s followed by two rinses in 70% ethanol for 10 s each. After washing in distilled water, staining was checked under the light microscope and the lithium carbonate incubation with following washing step repeated until a sharp contrast between grey and white matter was visible. Sections were then incubated in 0.1% cresyl violet staining solution for 4 min and thereafter again dehydrated using isopropyl alcohol and xylene and mounted Entellan (Sigma-Aldrich, St. Louis, MO, USA). b) PFA-fixed brain samples were obtained as previously reported [85]. Briefly, the animals were terminally anesthetized with ketamine and xylazine, transcardially perfused with a peristaltic pump (speed: 5 mL/min) by infusing 25 mL of ice-cold PBS followed by 50 mL of cold 4% PFA (pH 7.4). Brain samples were quickly dissected, post-fixed in 4% PFA for 18 h at 4 °C, washed in PBS, and dehydrated in 30% sucrose for 36 h. Samples were sectioned at -18 °C in a cryostat (Leica CM1950, Wetzlar, Germany) at a thickness of 30 μm. The sections were washed in PBS, blocked in PBS + 3% BSA + 0.5% Triton-X or PBS + 10% goatserum + 0.5% Triton-X and incubated with the appropriate antibody combination from 16–72 h at 4 °C, followed by washing in PBS (45 min × 3) and incubation with a corresponding combination of fluorophore-coupled secondary antibodies for 2 h at room temperature; after washing, sections were dried and mounted with Mowiol® (Carl Roth, Karlsruhe, Germany). For each experimental group or timepoint, at least three animals were processed and analyzed. Images were taken with an All-in-one fluorescence microscope BZ-X810 from Keyence (Neu-Isenburg, Germany) with a DAPI, FITC, TexasRed®, Cy5®, and a bright field filter, 10–40 × magnification, the BZ Viewer for capturing, and BZ Analyzer for merging. Images of stained sections were captured with equal exposure times per channel and the same graphical pre-processing within one experiment. For analysis and quantification, the ImageJ software (Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA) was used.
Antibodies for immunoblotting and immunostaining
The following antibodies were used for immunoblot analysis: rabbit-anti-IKK2 (CST-2678, Cell Signaling, Danvers, MA, USA), rabbit-anti-p-p65 (CST-3033, Cell Signaling, Danvers, MA, USA), rabbit-anti-p65 (sc-372, Dallas, TX, USA), chicken-anti-GFP (ab13970, Cambridge, UK), rabbit-anti-tubulin (ab6046, Cambridge, UK), rabbit-anti-PLP1 (CST-28702, Cell Signaling, Danvers, MA, USA), rabbit-anti-MOG (CST-96457, Cell Signaling, Danvers, MA, USA), rabbit-anti-MBP (CST-2396, Cell Signaling, Danvers, MA, USA), rabbit-anti-GAPDH (CST-3686, Cell Signaling, Danvers, MA, USA), rabbit-anti-p-eIF2α (CST-3597, Cell Signaling, Danvers, MA, USA), rabbit-anti-eIF2α (CST-5324, Cell Signaling, Danvers, MA, USA), rabbit-anti-ERK2 (sc-1647, Dallas, TX, USA).
For immunofluorescence, the following primary antibodies were used: mouse-anti-CC1 (OP80, Merck, Darmstadt, Germany), mouse-anti-GSTπ (BD610719, BD, Franklin Lakes, NJ, USA), chicken-anti-GFP (GFP-1020, AvesLab, Davis, CA, USA), mouse-anti-GFAP (sc-33673, Dallas, TX, USA), rabbit-anti-NG2 (AB5320, Merck, Darmstadt, Germany), rabbit-anti-MBP (Biolegend 836,504, San Diego, CA, USA), mouse-anti-Neurofilament-H (SMI32P) (Biolegend 801,701, San Diego, CA, USA), rabbit-anti-ß3-tubulin (Biolegend 802,001, San Diego, CA, USA), rabbit- anti RFP (abcam ab124754, Cambridge, UK).
RNA extraction, cDNA synthesis, and qRT-PCR
Total RNA from tissue samples and cell pellets was isolated using the Peq-Gold Trifast Kit (Peqlab, Erlangen, Germany) or RNA-Solv® Reagent (Omega Bio-Tek, Norcross, GA, USA) as described in the manufacturer’s protocol. 1 μg of total RNA was used to synthesize cDNA with the Transcriptor High Fidelity cDNA Synthesis kit (Roche, Grenzach-Whylen, Germany) with oligo-dT-primers according to the manufacturer’s instructions. Quantitative PCR (qPCR) assays were run in the Lightcycler 480 Instrument (Roche, Grenzach-Whylen, Germany) with primers and hydrolysis probes designed by the Roche Universal Probe Library (UPL) system. As a reference gene, the housekeeping gene hypoxanthine–guanine phosphoribosyl transferase (Hprt) was used. Primer sequences and UPLs are available upon request.
Electron microscopy
Brains were quickly dissected and fixed in 0.1% Glutaraldehyde (GA) (Agar Scientific, Stansted, UK), 4% PFA, 2% Sucrose in 0.1 M Sorensen Phosphate Buffer for 24 h. Area of interest (Corpus Callosum) was dissected sagittal and again fixed overnight in 2.5% GA, 0.1% Sucrose in 0.1 M Sorensen Phosphate Buffer. Samples were then washed in PBS and post-fixed in 2% osmium tetroxide in PBS. After dehydrating the samples in a graded series of isopropanol, they were blockstained in 2% uranyl acetate in ethanol and embedded in Epon. Semi-thin (500 nm) sections were stained with toluidine blue and analyzed using light microscopy to find the area of interest. Ultra-thin Sects. (80 nm) were cut on a microtome using a diamond knife (Leica EM UC7, Wetzlar, Germany) and collected on carbon coated formwar films on 200 mesh copper grids (Plano, Wetzlar, Germany) contrasted with 0.3% lead citrate for 1 min and imaged using a TEM 1400 (Jeol, Tokyo, Japan).
Cellular respiration and extracellular acidification
Primary OLs and microglia (see above) were seeded in XF96 V3 PS cell culture Microplates (Agilent Technologies, Santa Clara, USA) coated with 0.01% Poly-D-Lysin (Thermo Fisher Scientific, Waltham, MA, USA) at a density of 60.000 cells/well and 50.000 cells/well, respectively. OLs were plated in the following medium: NeuroMacs Medium supplemented with 2% NeuroBrew-21 (Miltenyi Biotec, Bergisch-Gladbach, Germany), 1% penicillin/streptomycin, 0.5 mM Glutamine (Thermo Fisher Scientific, Waltham, MA, USA). For proliferation, 10 ng/mL PDGF-AA and 10 ng/mL FGF-2 (Peprotech, Hamburg, Germany) were added freshly to the medium. After two days, 2 nM Triiodothyronine (T3) (Sigma-Aldrich, St. Louis, MO, USA) was added for differentiation and furthermore every second day until measurement at day 8. Microglia were seeded in DMEM supplemented with 10% FBS, 1% penicillin/streptomycin, 2 mM Glutamine (Thermo Fisher Scientific, Waltham, MA, USA). The medium was exchanged every second day until measurement at day 8. On the day of measurement, the cells were washed with XF assay medium (Agilent Technologies, Santa Clara, USA) containing 1 mM sodium pyruvate and 2 mM glutamine and incubated in fresh XF medium for 1 h under CO2 free conditions. Thereafter, oxygen consumption and extracellular acidification rates (OCR and ECAR) were measured simultaneously using a Seahorse XFe96 Flux Analyzer (Agilent Technologies, Santa Clara, CA, USA). The following injections and final concentrations were used: glucose (10 mM), oligomycin (2 μM), carbonylcyanide-p-trifluoromethoxyphenylhydrazone (FCCP, 2 μM) and antimycin A (0.5 μM)/ rotenone (0.5 μM). Data were normalized to JanusGreen as a surrogate for cell number per well.
Rna-sequencing analysis
Total RNA extraction was performed as stated above, library construction and RNA-sequencing were conducted by Novogene (London, UK). Messenger RNA was purified from total RNA using poly-T oligo-attached magnetic beads. After fragmentation, the first strand cDNA was synthesized using random hexamer primers, followed by the second strand cDNA synthesis using dUTP for directional library. The library was checked with Qubit and real-time PCR for quantification and bioanalyzer for size distribution detection. Quantified libraries were pooled and sequenced on Illumina platforms, according to effective library concentration and data amount. The clustering of the index-coded samples was performed according to the manufacturer’s instructions. After cluster generation, the library preparations were sequenced on an Illumina platform and paired-end reads were generated. Raw data (raw reads) of FASTQ format were firstly processed through fastp. In this step, clean data (clean reads) were obtained by removing reads containing adapter and poly-N sequences and reads with low quality from raw data. At the same time, Q20, Q30 and GC content of the clean data were calculated. All the downstream analyses were based on the clean data with high quality. Reference genome (GRCm38) and gene model annotation files were downloaded from genome website browser (NCBI/UCSC/Ensembl) directly. Paired-end clean reads were aligned to the reference genome using the Spliced Transcripts Alignment to a Reference (STAR) software, which is based on a previously undescribed RNA-seq alignment algorithm that uses sequential maximum mappable seed search in uncompressed suffix arrays followed by seed clustering and stitching procedure. STAR exhibits better alignment precision and sensitivity than other RNA-seq aligners for both experimental and simulated data. FeatureCounts was used to count the read numbers mapped of each gene. And then RPKM of each gene was calculated based on the length of the gene and reads count mapped to this gene. RPKM, Reads Per Kilobase of exon model per Million mapped reads, considers the effect of sequencing depth and gene length for the reads count at the same time, and is currently the most commonly used method for estimating gene expression levels [86].
Ex-vivo magnetic resonance imaging
Magnetic resonance imaging (MRI) was performed on a dedicated ultrahigh field 11.7 T small animal system (BioSpec 117/16, Bruker Biospin, Ettlingen, Germany) equipped with a 9 cm gradient insert (BGA-S9) operating with ParaVision 6.01. All data were acquired using a cryogenically-cooled 1H two-element surface (MRI CryoProbe™, Bruker BioSpec, Ettlingen, Germany) transmit/receive coil. For 3D brain imaging: FLASH with acquisition parameters as follows: TR/TE = 55/7 ms, flip angle FA = 15°, matrix 300 × 224 × 244, Δr = 40 × 40 × 40 µm3). field-of-view FOV = 12 × 9 × 9.85 mm3, and bandwidth = 50 kHz using 18 signal averages without interpolation. TA: 14h40m. Sequence weighting was T1-weighted.
Traumatic brain injury – Closed Head Injury model (CHI)
Twelve-week-old wild type or NF-κB reporter gene mice [46] were subjected to experimental CHI with a standardized weight-drop device [34, 87]. In brief, the animals were anesthetized with ketamine (Pfizer Pharma, Karlsruhe, Germany), with an i.p. dose of 100 mg/kg bodyweight, and 2% xylazine (Bayer Health Care, Monheim, Germany), with an i.p. dose of 16 mg/kg body weight Afterward, the skull was exposed by a longitudinal incision of the skin, and a focal blunt injury was induced in the left hemisphere by dropping a 330 g metal rod on the skull from a height of 2.7 cm. After trauma, the mice received supporting oxygenation with 100% O2, the wound was sutured, and the animals were placed into a warmed recovery cage with ad libitum access to food and water. Buprenorphine analgesia (Temgesic; Essex Pharma, Munich, Germany) was administered subcutaneously (0.03 mg/kg body weight) immediately after trauma and every 8 h thereafter until 24 h.
Statistical analysis
Statistical analyses were performed with Prism software (GraphPad, San Diego, CA, USA) and are indicated in the specific figure legend. One- or 2-way ANOVA with Bonferroni’s correction was used to compare independent measurements at one or different time points, respectively. For nonparametric analysis Mann–Whitney U was used. All data are shown as mean ± SEM, statistical significance was set at P < 0.05.


留言 (0)