Strains, media, and transformations
The A. nidulans strains used in this study are listed in Table S1. The strains were grown in a minimal medium (MMG) or a complete medium (YG) (Fukuda et al. 2009; Jin et al. 2021). When using a solid medium, 1.5% (w/v) agar was added. MMG or YG medium was supplemented with 0.5 mg/ml pyridoxine, 1.12 mg/ml uracil, 2.44 mg/ml uridine when necessary, and the additives were indicated by initials in lowercase letters after the medium name. The transformations of A. nidulans and Escherichia coli, and the propagation of E. coli were conducted as described previously (Tsuizaki et al. 2013).
Construction of strains, plasmids and DNA fragments
The primers used in this study are listed in Table S2.
The 3FLChB strain, in which 3xFLAG-tagged ChsB was endogenously produced under the chsB promoter, was constructed as follows: A 1.5-kb fragment was amplified from the total DNA of the GPChBP strain using primers ‘5′-YFP-chsB for’ and ‘3xFLAG-chsB-1 rev.’ An 8.1-kb fragment was amplified from the total DNA of the GPChBP strain using primers ‘3xFLAG-chsB-2 for’ and ‘3′-YFP-chsB rev.’ The fragments were fused via PCR using primers ‘5′-YFP-chsB for’ and ‘3′-YFP-chsB rev’ to yield a 9.6-kb fragment. The 9.6-kb fragment was transformed into the A1149 strain. The transformants in which the gene encoding 3xFLAG-tagged ChsB was introduced into the 5′-terminus of chsB were selected using Southern blot analysis and designated as 3FLChB (Fig. S2A). The 3FLChBP strain, in which the wild-type pyrG was introduced into the 3FLChB strain, was constructed as follows: A 1.9-kb fragment was amplified from the total DNA of the A26 strain using primers ‘pyrG ORF-1 kb for’ and ‘pyrG ORF rev.’ The 1.9-kb amplified fragment was transformed into the 3FLChB strain. The transformant was designated as 3FLChBP.
A plasmid, pegfp–chsB, was constructed as follows: A 9.0-kb fragment was amplified from the total DNA of GPChBP strain using primers ‘5′-9xHA-chsB for’ and ‘3′-9xHA-chsB rev.’ The amplified fragment and pBlueScript II SK( +) were digested with Hind III and ligated to yield pegf–chsB.
The GPChBPΔ1–20, GPChBPΔ1–40, GPChBPΔ1–60, GPChBPΔ1–80, or GPChBPΔ1–100 strain that produced mutant ChsB with the deletion of the 1st–20th, 1st–40th, 1st–60th, 1st–80th, or 1st–100th amino acids at the N-terminus, respectively, instead of wild-type ChsB was constructed as described subsequently. A 2.1-kb fragment was amplified from the total DNA of GPChBP strain using primers ‘egfp-chsB-F’ and ‘3′-egfp rev’. DNA fragments were amplified from pegfp–chsB using primers ‘egfp-chsB-Δ1-20–2-F’ and ‘egfp-chsB-R’ for GPChBPΔ1–20; ‘egfp-chsB-Δ1-40–2-F’ and ‘egfp-chsB-R’ for GPChBPΔ1–40; ‘egfp-chsB-Δ1-60–2-F’ and ‘egfp-chsB-R’ for GPChBPΔ1–60; ‘egfp-chsB-Δ1-80–2-F’ and ‘egfp-chsB-R’ for GPChBPΔ1–80; and ‘egfp-chsB-Δ1-100–2-F’ and ‘egfp-chsB-R’ for GPChBPΔ1–100. Each fragment was then fused with the 2.1-kb fragment. Each of the obtained DNA fragments was transformed into the A1149/pyrG-1 strain (Katayama et al. 2012). The transformants in which the wild-type chsB was replaced with the introduced fragment were selected using Southern blot analysis and designated as GPChBPΔ1–20, GPChBPΔ1–40, GPChBPΔ1–60, GPChBPΔ1–80, and GPChBPΔ1–100, corresponding to the deleted regions of the ChsB N-termini.
The GPChBPΔ1–115 or GPChBPΔ1–140 strain that produced the mutant ChsB with the deletion of the 1st–115th or 1st–140th amino acids at the N-terminus, respectively, instead of wild-type ChsB, was constructed as follows: An 11.5-kb or 11.4-kb fragment was amplified from pegfp–chsB using primers ‘egfp-chsB-Δ1-115-F’ and ‘3′-egfp rev’ for GPChBPΔ1–115 or ‘egfp-chsB-Δ1-140-F’ and ‘3′-egfp rev’ for GPChBPΔ1–140, respectively. The 11.5-kb or 11.4-kb fragment was self-ligated to yield pegfp–chsB-Δ1–115 or pegfp–chsB-Δ1–140, respectively. The pegfp–chsB-Δ1–115 or pegfp–chsB-Δ1–140 strain was digested with Hind III to yield an 8.6-kb or 8.5-kb fragment, respectively. Each fragment was transformed into the A1149/pyrG-1 strain. The transformants in which the wild-type chsB was replaced with the introduced fragment were selected using Southern blot analysis and designated as GPChBPΔ1–115 or GPChBPΔ1–140, corresponding to the deleted regions of the ChsB N-termini.
The GPChBPΔ21–40, GPChBPΔ41–60, GPChBPΔ61–80, GPChBPΔ81–100, GPChBPΔ101–115 and GPChBPΔ116–140 strains that produced mutant ChsBs with the deletion of 1–20, 21–40, 41–60, 61–80, 81–100, 101–115, and 116–140 amino acids at the N-terminus, respectively, instead of wild-type ChsB, were constructed as described subsequently. DNA fragments encoding the N-terminal regions of ChsB were amplified from the total DNA of GPChBP strain using primers ‘egfp-chsB-F’ and ‘egfp-chsBΔ21-40–1-R’ for GPChBPΔ21–40; ‘egfp-chsB-F’ and ‘egfp-chsBΔ41-60–1-R’ for GPChBPΔ41–60; ‘egfp-chsB-F’ and ‘egfp-chsBΔ61-80–1-R’ for GPChBPΔ61–80; ‘egfp-chsB-F’ and ‘egfp-chsBΔ81-100–1-R’ for GPChBPΔ81–100; ‘egfp-chsB-F’ and ‘egfp-chsBΔ101-115–1-R’ for GPChBPΔ101–115; and ‘egfp-chsB-F’ and ‘egfp-chsBΔ101-140–1-R’ for GPChBPΔ116–140. DNA fragments encoding the C-terminal regions of ChsB were amplified from pegfp–chsB using primers ‘egfp-chsB-Δ21-40–2-F’ and ‘egfp-chsB-R’ for GPChBPΔ21–40; ‘egfp-chsB-Δ41-60–2-F’ and ‘egfp-chsB-R’ for GPChBPΔ41–60; ‘egfp-chsB-Δ61-80–2-F’ and ‘egfp-chsB-R’ for GPChBPΔ61–80; ‘egfp-chsB-Δ81-100–2-F’ and ‘egfp-chsB-R’ for GPChBPΔ81–100; ‘egfp-chsB-Δ101-115–2-F’ and ‘egfp-chsB-R’ for GPChBPΔ101–115; and ‘egfp-chsB-Δ115-140–2-F’ and ‘egfp-chsB-R’ for GPChBPΔ116–140. Each of the N-terminal and C-terminal fragments was fused using fusion PCR with primers ‘egfp-chsB-F’ and ‘egfp-chsB-R.’ Each obtained fragment was transformed into the A1149/pyrG-1 strain. The transformants in which the wild-type chsB was replaced with the introduced fragment were selected via Southern blot analysis and designated as GPChBPΔ21–40, GPChBPΔ41–60, GPChBPΔ61–80, GPChBPΔ81–100, GPChBPΔ101–115 or GPChBPΔ116–140, corresponding to the deleted regions of the ChsB N-termini.
Southern blot analysis
Genomic DNA of the transformed strains was digested with the restriction enzymes listed in Figure S2, and transferred to Nylon Membrane, positively charged (Roche, Mannheim, Germany). Probes were amplified using PCR DIG Labeling MixPLUS (Roche) according to the manufacturer’s instructions. The probes were detected using Anti-Digoxigenin-AP (Roche) and CDP-Star™ detection reagent (GE Healthcare, IL, USA) according to the manufacturer’s instructions.
Preparation of cell lysate
Total cell lysates were extracted as previously described (Jin et al. 2021). To determine the phosphorylation level of ChsB, urea was added to the lysate to a final concentration of 6 M, and the lysate was incubated at 100 °C for 5 min before performing SDS–polyacrylamide gel electrophoresis.
Immunoprecipitation
Protein G FG beads (0.1 mg; TAS8848N1173; Tamagawa Seiki, Nagano, Japan) were washed with phosphate-buffered saline (PBS) (0.14 M NaCl, 8 mM Na2HPO4, 2.7 mM KCl, 1.5 mM KH2PO4) twice. PBS (10 μl) and 5 μl monoclonal anti-FLAG antibody produced in rabbit (F7425; Merck Millipore, MA, USA) or anti-GFP (118144600001; Roche) were added to the beads and centrifuged at 1400 rpm for 30 min at room temperature. The beads were washed three times with wash buffer (1 mM ethylenediaminetetraacetic acid [EDTA], 10% glycerol [v/v], 10 mM 4-[2-hydroxyethyl]-1-piperazineethanesulfonic acid sodium hydroxide [HEPES–NaOH; pH 7.9], and 50 mM KCl). Wash buffer (20 μl) and 200 μl KCl buffer (0.4 mM CaCl2, 0.4 mM EDTA and 20% glycerol [v/v], 40 mM HEPES–NaOH [pH 7.9], 300 mM KCl, 2 mM MgCl2, 0.2% NP-40 [w/v] and 0.4 mM phenylmethylsulfonyl fluoride) were added to the beads with agitation. The beads were magnetically separated after spin-down, and the supernatant was removed. The beads were washed twice with KCl buffer. The cell lysate was diluted to 1 mg/ml with KCl buffer, and 200 μl of the diluted lysate was added to the beads with agitation. After agitation at 4 °C for 2 h, the beads were washed three times with KCl buffer. 0.1 M Glycine–HCl buffer (28 μl; pH 2.5) was added to the beads with agitation, after which the beads were placed on ice for 5 min. After magnetic separation, the supernatant was collected and 2 μl of Tris–HCl buffer (pH 9.0) was added to obtain an immunoprecipitation fraction.
To purify EGFP-fused ChsBs, 500 µl of total cell lysate (protein concentration: 5 mg/ml) was added to 12.5 µl of GFP-Trap Magnetic Agarose (Proteintech Group, IL, USA). Bead equilibration, protein binding, washing, and elution with acidic elution buffer were conducted as described in the manufacturer’s documentation. The elution buffer was 50 µl, and the elution was performed twice.
Phosphatase treatment
To dephosphorylate proteins, 3 μl of 10 × alkaline phosphatase buffer (Takara Bio, Shiga, Japan), 25 μl of calf intestine alkaline phosphatase buffer (CIAP; 1 mM MgCl2 and 50 mM Tris–HCl [pH 7.5]) and 3 μl of CIAP (Takara Bio) were added to 2 μl of the immunoprecipitation fraction and incubated at 37 °C for 2 h. Phosphatase inhibitor cocktail 1 (1 μl, Merck Millipore) and phosphatase inhibitor cocktail 2 (1 μl, Merck Millipore) were also added to the solutions.
To dephosphorylate EGFP-fused ChsBs, 10 µl of the elution fraction, 3 µl of 10 × Alkaline phosphatase buffer (Takara Bio), 2 μl of Alkaline Phosphatase (Calf intestine) (Takara Bio), and 15 µl of distilled water were mixed and incubated at 37 °C for 2 h.
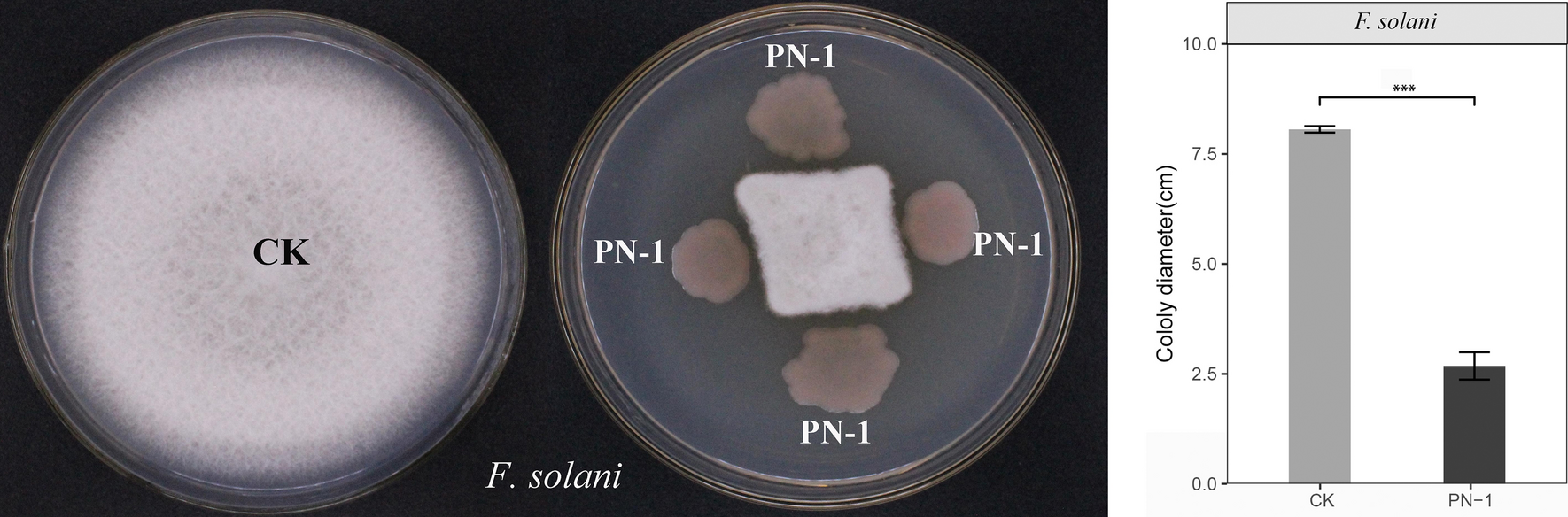
Measurement of colony area
The area of the colony was measured using Fiji (https://imagej.net/software/fiji/).
Fluorescence microscopy and measurement of fluorescence intensity
Fluorescence microscopy and measurement of fluorescence intensity were conducted as previously described (Jin et al. 2021).
CFW and CMAC staining
To observe the septa, the hyphae were treated with CFW solution (0.5 mg/mL fluorescent brightener 28 (F-3543; Sigma-Aldrich, MO, USA), 5% [w/v] potassium hydroxide, 10% [v/v] glycerol) for 1 min at room temperature. To visualize vacuoles, the hyphae were treated with 100 µM 7-amino-4-chloromethylcoumarin-L-alanyl-L-proline amide (CMAC-Ala-Pro; Y7531; Thermo Fisher Scientific, MA, USA) in MMG liquid medium for 30 min at room temperature in the dark. Fluorescence was observed using a U-MWU2 filter cube (Evident, Tokyo, Japan).
Estimation of protein concentration
The protein concentration of the cell lysate was determined using a Protein Assay BCA kit (Fujifilm, Tokyo, Japan) according to the manufacturer’s instructions. Absorbance at 562 nm was measured using a UV-1900 spectrophotometer (Shimadzu, Kyoto, Japan).
Immunoblot analysis
Immunoblot analysis was performed as described previously (Jin et al. 2021). Primary antibodies for β-tubulin, FLAG-tag, and GFP used in this study were monoclonal anti-β-tubulin antibody (clone: 10G10; Fujifilm), monoclonal anti-FLAG M2 antibody (F1804; Sigma-Aldrich), and monoclonal anti-GFP antibodies (clones: 7.1 and 13.1; 11814460001; Roche), respectively. Secondary antibodies were anti-mouse IgG and HRP-linked Antibody (7076S; Cell Signaling Technology, MA, USA). Chemiluminescence signals produced by ImmunoStar Zeta (Fujifilm) or SuperSignal West Atto Ultimate Sensitivity Substrate (Thermo Fisher Scientific) were detected using a CCD camera system (iBright FL1500 Imaging System; Thermo Fisher Scientific). Band intensities were quantified using Fiji Gel Analyzer.
Statistical analysis
Statistical analysis was performed using R (https://www.r-project.org).









留言 (0)