AnimalsEthical approval
All animal experiments were reviewed and approved by the Ethical Committee for Animal Experiments of KU Leuven, Belgium (P102/2017).
Housing
All mice (C57Bl/6 J, 8–12 weeks old) were housed with a maximum of 5 animals per cage under controlled standard conditions with ad libitum access to food pellets and tap water.
Progesterone-induced uterine gland knockout (PUGKO) mice model
Female and male C57Bl/6 J mice of 8–12 weeks old were mated. Litters derived from these couples were randomly assigned to the control group or treatment group. Pups assigned to the treatment group received daily progesterone injections (P4, 50 µg/g body weight in corn oil) from postnatal day 2 until postnatal day 10 to inhibit uterine gland formation. Pups assigned to the control group were injected with similar volumes of vehicle (corn oil) as the treatment group. Animals were used for experiments at the age of 8–12 weeks.
Conditional progesterone specific GCamp3 (F/F) mice
Mice genetically encoded with the Ca2+ sensor GCamp3 after a floxed stop codon in the Rosa26 locus were crossed with the progesterone receptor (PR)-cre mice that express the cre under the PR promoter. The crossing with a cre-line removes the stop codon and allows for the expression of the Ca2+ sensor in all tissues expressing the cre. Thus, in the offspring of these mating, the GCamp3 Ca2+ sensor will be present in all tissues expressing PR, amongst which the uterine epithelium.
Cell cultureMouse endometrial epithelial and stromal cells
The isolation of mouse endometrial epithelial (mEEC) and stromal (mESC) cells was performed as previously described [14]. Briefly, four to five female mice (C57Bl/6 J, Janvier France) were placed on male bedding the day before isolation to synchronize their estrous cycle. At the time of isolation, the estrous cycle phase was determined via vaginal smear examination. The mice were sacrificed whereafter the uteri were removed and placed in ice cold Hank’s Balanced Salt Solution 1X (Gibco, Thermo Fischer Scientific, Belgium) supplemented with 2% penicillin–streptomycin (Gibco, Thermo Fischer Scientific, Belgium) (HBSS +). After removal of fat and mucus, the uteri were cut open longitudinally and incubated for 1 h at 4 °C, 45 min at room temperature (RT) and 15 min at 37 °C in HBSS + supplemented with 2.5% pancreatin (Sigma-Aldrich, Belgium) and 0.25% trypsin (from bovine pancreas, Sigma Aldrich, Belgium). Thereafter, the uteri were vortexed and rinsed in HBSS + for 3 consecutive times before all solutions were pipetted onto a 100 µm cell strainer (BD Falcon, Fisher Scientific, France) in order to obtain the epithelial sheets. The cell suspension was centrifuged at 500 × g for 5 min and ultimately seeded onto the appropriate cell culture plates. After isolation of mEEC, the uteri were further digested in 0.05% Trypsin–EDTA solution (Gibco, Thermo Scientific, Belgium) supplemented with 0.1 mg/ml type IA collagenase (Sigma-Aldrich, Belgium) for 30 min on a shaker at 37 °C. After digestion, the solution was gently shaken for 10 s to release the stromal cells. The uteri were further rinsed in HBSS + , transferred to mESC growth medium and shaken. This was repeated for 3 consecutive times. Cells were finally seeded onto the desired cell cultures plates and cultured in Dulbecco modified Eagle’s medium (DMEM)/F12 (Gibco, Thermo Scientific, Belgium) supplemented with 10% fetal bovine serum (FBS), (Gibco, Thermo Scientific, Belgium), 0.5 µg/ml amphotericin B (Gibco, Thermo Scientific Belgium) and 100 µg/ml gentamicin (Gibco, Thermo Scientific, Belgium).
Human endometrial organoids
Human endometrial organoids of healthy women (hEMO) were cultured as described by Boretto et al. [15]. Organoids were exposed to hormonal treatments mimicking the uterine microenvironment during the menstrual cycle [15, 16]. The following protocol was used: 2 days endometrial medium (no hormonal supplement), followed by a supplement of β-estradiol (E2; 10 nM) for 2 days to induce the proliferative phase and finally 4 days of EPC (E2 (10 nM) + progesterone (P4; 1 µM) + cAMP (0.5 mM)) to induce the mid-secretory phase (including the window of implantation) [16, 17].
RNA extraction and RT-qPCR
Total RNA was extracted from mEEC and hEMO using the RNeasy mini kit (Qiagen, The Netherlands) according to the manufacturers’ guidelines. RNA concentration and quality were evaluated using the Nanodrop method (Isogen Life Science, Belgium). cDNA was generated from 1 µg total RNA using the First-Strand cDNA Synthesis Kit (GE Healthcare, Belgium). RT-qPCR was performed using specific TaqMan gene expression assays (Life Technologies, Belgium) in the StepOne PCR system (Applied Biosystems, Belgium). Phosphoglycerate Kinase 1 (Pgk1) and TATA-Box Binding Protein (Tbp) were used as endogenous controls for the analysis of gene expression in mEEC. For the hEMO, the endogenous controls were hypoxanthine phosphoribosyl transferase 1 (HPRT1) and phosphoglycerate kinase 1 (PGK1). Data are represented as mean ± SEM of 2(−ΔCt) for which ΔCt = Ctgene of interest – Ctgeometric mean of endogenous control. All experiments were performed on triplicate cDNA samples.
RNAscope in situ hybridization
In situ hybridization (ISH) of Par-2 (F2rl1), E-cadherin (Cdh1), Trpv6 and Enac (Scnn1a) on isolated EEC, hEMO, uterine and kidney tissue were performed using the RNAscope Multiplex Fluorescent Reagent Kit (Advanced Cell Diagnostics, US). All ISH assays were carried out according to the manufacturers’ guidelines for formalin-fixed, paraffin embedded samples. Images were taken by the use of a fluorescence microscope (Nikon Eclipse Ci-E) with constant gain and exposure times at a 10 × or 20 × magnification for all images.
Immunohistochemistry
FOXA2 immunostaining was performed on 4 µm cross-section of paraffin-embedded uterine tissue derived from 8–12 weeks old control and PUGKO animals. The sections were deparaffinized in xylene and rehydrated in graded alcohol series. Antigen retrieval was performed by boiling the uterine sections in 10 mM citrate buffer (pH 6) for 60 min. After a block with 3% normal goat serum, the sections were incubated overnight with primary rabbit monoclonal anti-FOXA2 antibody (2 µg/ml; Abcam ab108422). Afterwards, the tissues were rinsed in TRIS-buffered saline (TBS) and peroxidase-labeled secondary goat anti-rabbit antibody was used to localize FOXA2. Finally, the sections were counterstained with Maeyer Hematoxylin before mounting.
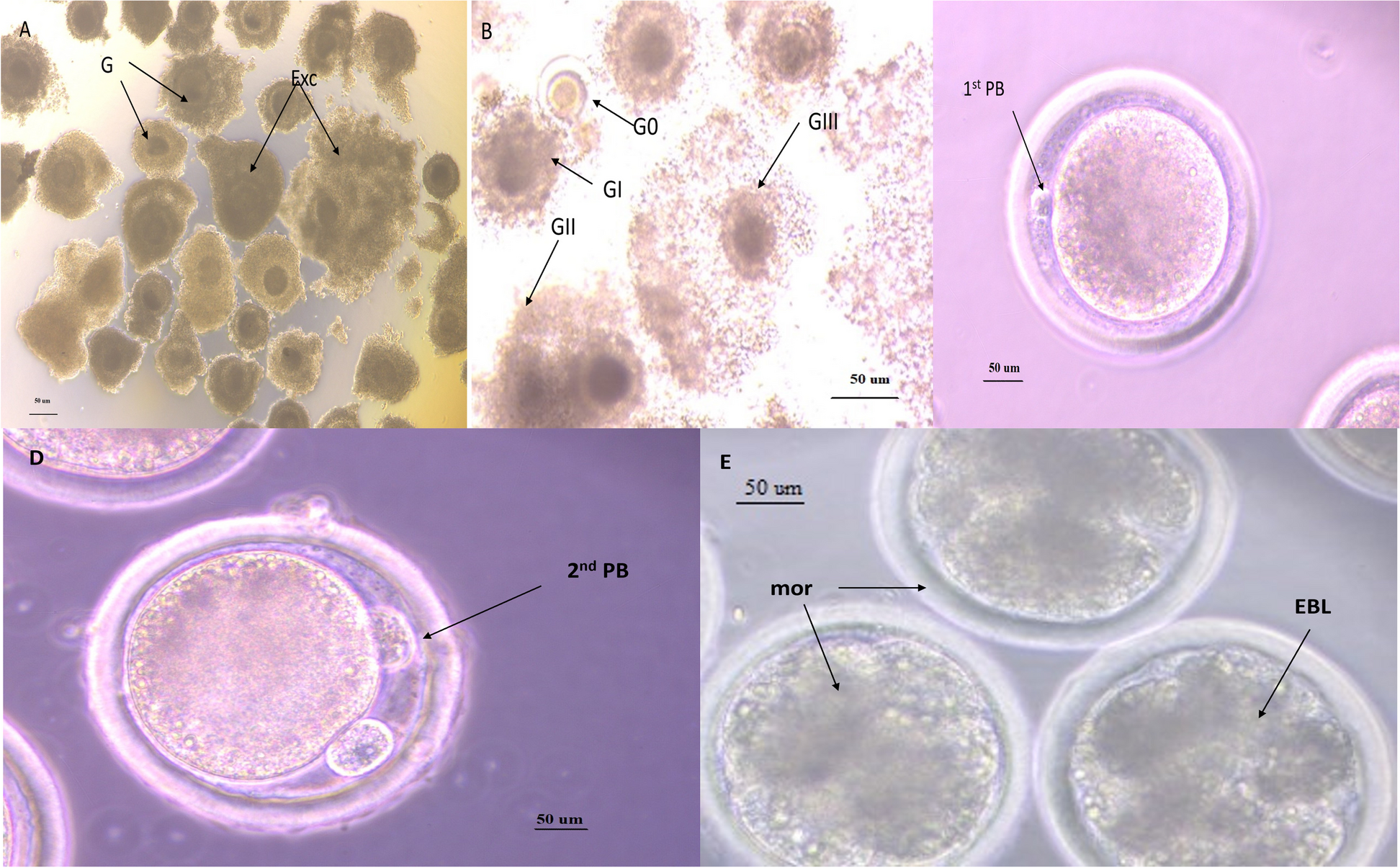
Human blastocysts (day post fertilization (dpf) 5 and dpf6) were collected at the Leuven University Fertility Centre with approval of the Ethical Committee of the UZ/KU Leuven (S62765) and the federal Commission for medical and scientific research on embryo’s in vitro, after written informed consent of the patient. Intact human blastocysts were individually fixed in 4% paraformaldehyde (PFA) for 20 min at RT. After fixation, samples were washed in 3% bovine serum albumin (BSA)/ phosphate buffered saline (PBS) and permeabilized with 0.5% Triton X-100 (T-8787, Sigma-Aldrich, USA) for 20 min at RT. The blastocysts were incubated in 10% FBS/PBS blocking solution for 30 min at RT and subsequently incubated with primary Anti-Trypsin (D-1) antibody (sc-137077) or PBT control (0.1% Triton + 3% FBS) in 10% BSA/PBS overnight at 4 °C in the dark. Blastocysts were washed in 3% BSA/PBS before incubation with secondary antibody goat anti-mouse Alexa Fluor 647 (Abcam ab150115) for 1.5 h at RT in the dark. Samples were washed in 3% BSA/PBS before incubation with HOECHST (10 nM; 33,342; Thermo Scientific) for nuclear staining. Samples were again washed in 3% BSA/PBS and finally mounted on glass bottom dishes (Cellvis). Confocal scanning microscopy was used to obtain fluorescent images. A Nikon Ti2 inverted AX R microscope was used in combination with a 20 × Plan Apo VC objective lens. The setup was controlled by NIS-Elements (NIS 5.40, Nikon Instruments Europe). DAPI and far red were respectively excited with 405 and 647 nm and the emission was collected with 429-474 nm and 662-737 nm filters. For post processing, NIS-Elements (5.40, Nikon Instruments Europe) was used. The intensity of the z-stacks was equalized (in z) via histogram stretching and the images were denoised using denoise.ai.
RNA sequencingBulk RNA sequencing
Three independent isolations were used for these experiments. After mEEC were isolated according to abovementioned protocol, cells were incubated with trypsin (2 µg/ml) or control medium and collected at 0 h (baseline), 12 h and 24 h after incubation. Cells were collected in lysis buffer (RNeasy mini kit, Qiagen) and stored at -80 °C until further processing. Total RNA was extracted using the RNeasy mini kit (Qiagen, The Netherlands), according to the manufacturers protocol. The quality was evaluated using the Nanodrop method (Isogen Life Science, Belgium). The sequencing libraries were prepared with the Illumina TruSeq Stranded mRNA sample preparation protocol according to the manufacturers’ guidelines and sequenced on an Illumina NovaSeq6000 at the Nucleomics Core, VIB, Belgium. The reads were aligned to the GRCm38 mouse genome assembly (GENCODE release M25 [18]) using version 2.7.1 of the STAR software [19], and the transcript abundance was quantified as TPM (transcripts per million) with RSEM 1.3.1 [20].
Single cell sequencing
Isolated mEEC from control and PUGKO samples were mechanically processed to obtain single cells and resuspended in 0.04% BSA in PBS. Library preparation was performed using the Chromium-based single cell 10X Genomics platform according to the manufacturers protocol (10 × Genomics, single cell RNA sequencing 3′, Chromium v2). Afterwards, samples were sequenced on an Illumina HiSeq (mean reads per cell: ~ 100.000—125.000) and processed using the Cell Ranger pipeline (10X Genomics) and Seurat program for single cell genomics (Satijalab).
Single cell sequencing data of the human endometrium were obtained via publicly available servers at www.reproductivecellatlas.orgwww.reproductivecellatlas.org. Gene expression was evaluated within the Seurat program for single cell genomics (Satijalab).
Mouse endometrial single cell sequencing data were obtained via publicly available servers at Gene Expression Omnibus (GEO). Gene expression was evaluated within the Seurat program for single cell genomics (Satijalab).
Functional measurementsPharmacology
Trypsin (from porcine pancreas; 2 µg/ml), elastase (from porcine pancreas; 3U/ml), aprotinin (20 µg/ml), amiloride (10 µM) and nifedipine (10 µM) were purchased from Sigma-Aldrich, Belgium. The selective PAR2 activator, 2-furoyl-LIGRLO-NH2 (5 µM), store operated calcium entry (SOCE) inhibitor YM58483 (1 µM) and Phospholipase C (PLC) inhibitor U73122 (5 µM) with its negative control U73343 (5 µM) were obtained from Tocris, Bioscience, UK. The specific PAR2 inhibitor I-191 (100 nM) was purchased at Axon Medchem, The Netherlands. Ionomycin (2 µM) was used as a positive control at the end of each experiment. All stock solutions were prepared in Milli Q water, DMSO or EtOH according to the manufacturers’ guidelines.
Calcium microfluorimetry
The calcium measurements were performed as previously described [21]. Absolute calcium concentrations were calculated from the ratio of the fluorescence signals at both wavelengths (F340/F380) after correction for the individual background fluorescence signals, using the Grynkiewicz Equation [22]:
$$\left[}^\right]= }_} \frac- }_}}_-\mathrm}$$
for which the calibration constants R0, R1 and Keff were determined as followed: R0 defines the ratio in Ca2+ free medium supplemented with 10 mM EGTA, whereas R1 comprises the ratio in high Ca2+ medium (10 mM). Keff, the effective binding constant, includes R0, R1, the dissociation constant of indicator dye KD, and the isocoefficient α, according to the following equation:
$$}_}= }_} \frac}_+ \mathrm}}_+ \mathrm}$$
The KD of Fura-2 and the isocoefficient α were assumed as described by Zhou and Neher [23]. Cells were considered responders if the amplitude of the rise in intracellular calcium during agonist application exceeded 100 nM and when the highest value of the derivative of the calcium trace during the application of an activator exceeded at least 3 times the standard deviation (SD) of the derivative during basal conditions. Oscillating cells were defined as cells showing at least 2 peaks of intracellular calcium increase, with at least one peak exceeding 100 nM and/or cells displaying a visually obvious oscillatory pattern (≥ 4 peaks). Frequency was calculated based on the number of calcium peaks divided by the time of stimulus application. Only cells that responded to the positive control, ionomycin, at the end of the experiment were taken into account. The following bath solution was used for all measurements (in mM): 150 NaCl, 2 CaCl2, 1 MgCl2, 10 D-glucose, 10 HEPES, pH 7.4 with NaOH. In calcium-free conditions, CaCl2 was omitted, MgCl2 increased to 2.5 mM, and the solution was supplemented with 5 mM EDTA. In sodium-free conditions, NaCl was replaced with 150 mM CsCl. All data points originate from at least three independent experiments (n ≥ 3).
Ex vivo calcium imaging
Female C57Bl/6 J mice between 8–12 weeks old were placed on male bedding the day before the experiment to synchronize their cycle phase. Prior to sacrifice, the cycle phase was determined via vaginal smear cytology [24]. Animals were sacrificed and uterus collected in cold (4 °C) synthetical interstitial fluid (SIF) solution. The SIF solution contained (in mM): 125 NaCl, 26.2 NaHCO3, 1.67 NaH2PO4, 3.48 KCl, 0.69 MgSO4, 9.64 D-gluconic acid, 5.55 D-glucose, 7.6 Sucrose and 2 CaCl2. The pH was buffered with carbogen gas to pH 7.4. The collected uteri were opened longitudinally and each horn was cut in half. Next, the tissue fragment was incubated in SIF solution at 4 °C to rest for at least one hour. For image acquisition, tissue fragments were fixed with the inner uterus lining towards the objective in a glass-bottom microwell dish (MatTek, 35 mm petri dish, Ashland, USA) and incubated in SIF solution supplemented with the chemical compound of interest. Image acquisition was performed using a Nikon NiE microscope equipped with a Yokogawa CSU-X spinning-disk module with dual camera (Andor iXon3) in combination with a Plan Fluor 10 × air objective (NA 0.30, 16 WD). The images were adjusted for movement using Fiji ImageJ (Linear Stack Alignment with SIFT plugin) and regions of interest were selected. Raw fluorescent traces were extracted and converted to ΔF/F0, for which the fluorescence at any given point is represented by F, and F0 is the background fluorescence.
Whole-cell path clamp experiments
Patch clamp recordings were measured with an EPC-10 amplifier and PatchMasterPro Software (HEKA Elektronik, Lambrecht, Germany). Current measurements were performed at a sampling rate of 20 kHz and currents were digitally filtered at 2.9 kHz. The pipette solution contained (in mM): 100 CsAsp, 45 CsCl, 10 EGTA, 10 HEPES, 1 MgCl2 (pH 7.2 with CsOH). The extracellular solution contained (in mM): 150 NaCl, 10 HEPES, 10 Glucose, 2 CaCl2, 1 MgCl2, (pH 7.4 with NaOH). The standard patch pipette resistance was between 2 MΩ and 4 MΩ when filled with pipette solution. 50–70% of the series resistance was compensated during the recordings. The applied voltage step protocols are described in the corresponding figure legends.
Data analysis and statistics
Calcium imaging and electrophysiological data were analyzed using IgorPro 6.2 (WaveMetrics, USA), WinASCD (Guy Droogmans, Leuven, Belgium) and OriginPro 8.6 (OriginLab Corporation, USA). RNA seq data was analysed in RStudio 1.4.1717 (RStudio, PBC). OriginPro 8.6 was further used for data display. Statistics were performed using GraphPad Prism 8.4.3 for Windows (GraphPad Software, USA). Statistical significance was considered when p < 0.05.

留言 (0)