
記住我
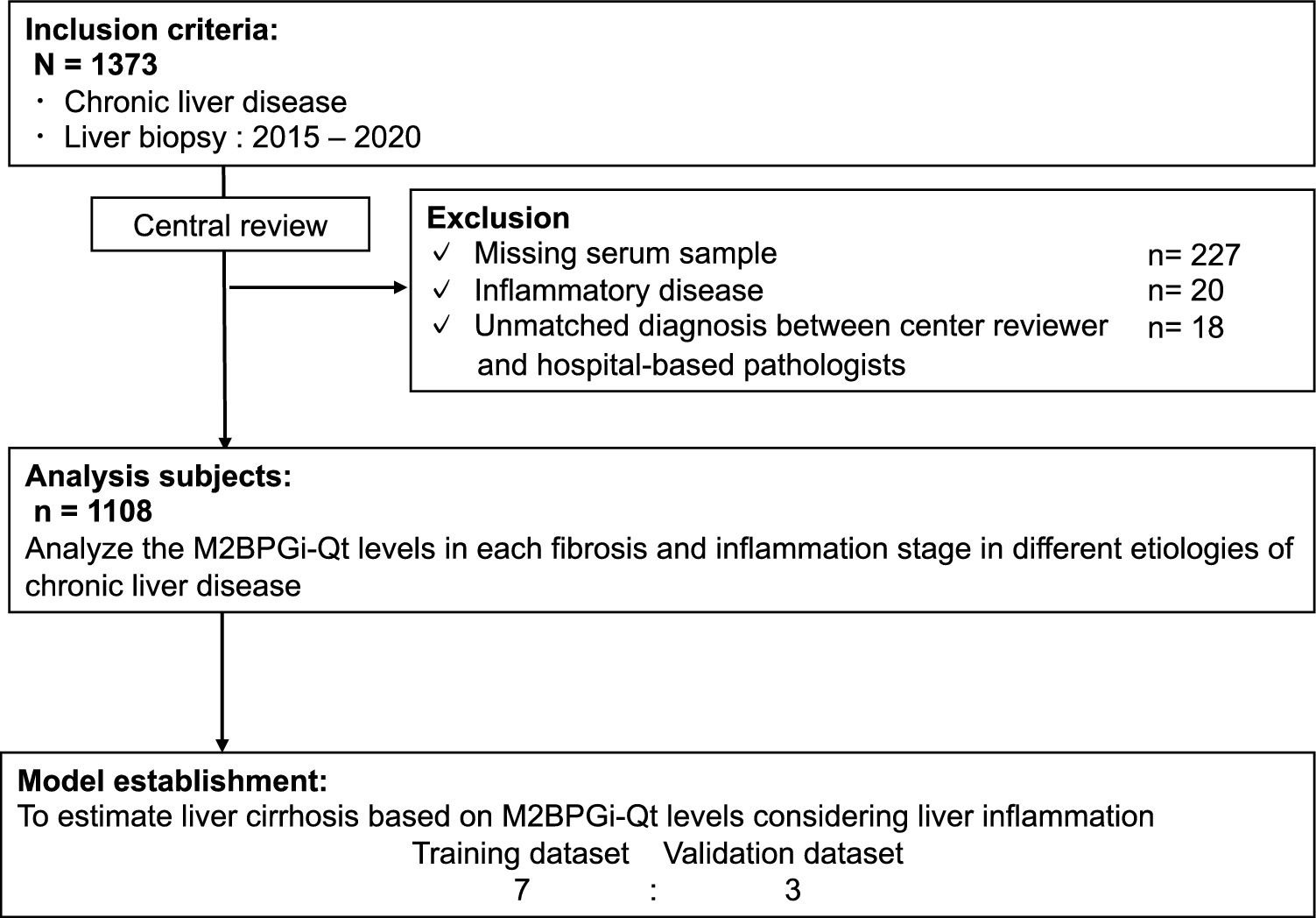


In current study, we sequenced 30 unique bulk tumor samples (matched 8 primary tumor samples and 22 liver metastatic samples) from 8 patients with CCLM, the primary and metastatic tumors were resected at the same period and no patient received chemoradiotherapy pre-operation. All tumors were microsatellite stable, and other characteristics information about the patients and samples are provided in Table 1. The collected samples were sequenced using whole-exome sequencing, and the workflow was illustrated in Fig. 1: firstly, samples from each of the eight patients were shown (Fig. 1a), and the specific locations of the primary and metastasis loci were shown in Table 1. Next, the HE stain of the primary and multiple metastases were underwent review, followed by next-generation whole-exome DNA sequencing, and the CC08 HE stains were depicted in Fig. 1b as an example. Finally, a series of data analyses were completed, including somatic mutations illustration; intra- and inter-tumor heterogeneity illustration by PyClone, evolution mode reconstruction as well as biological function assay.
Table 1 Patients and samples characteristicsFig. 1
Overview of patient cohort, samples, and study design. a Schematic diagram of the primary and paired multiple liver metastases locations for all eight patients. b Identification of pathology of colon primary and matched multiple metastasized liver tumor tissues, sequencing and clonal evolution analysis
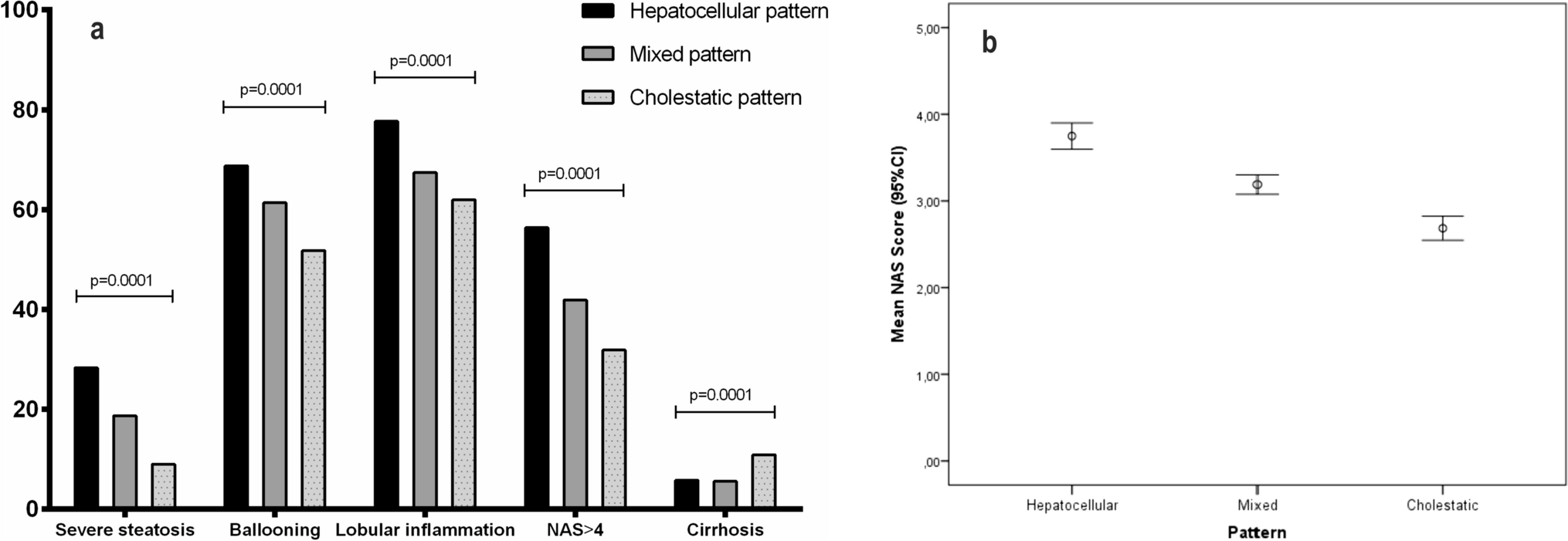
Diversity between primary and metastases in treatment-naive CCLMAn individual mutation profile was generated for each patient to present the genetic homogeneity and heterogeneity between primary and metastatic lesions (supplement materials S1). As shown in Fig. 2a and supplement materials S1, “Private” and “Shared” stood for heterogeneity, whereas “Ubiquitous” stood for homogeneity, evidently, mutations differed significantly between primary lesions and metastases, as well as among multiple metastases in all eight patients.
Fig. 2
Comparison of somatic mutations in treatment-naïve colon cancer between primary and paired multiple metastases. a Heatmaps show the distribution of all nonsynonymous mutations. Presence (yellow) or absence (blue) of each mutation is marked for each sample within one individual CC01. Private is sample specific (red), shared means mutations occurring in more than one tissue (brown) and ubiquitous is mutations identified in each tumors (green). The mutated genes are listed in the supplementary materials. b Oncoplot showing the TMB in each sample (top panels); somatic mutation landscapes of the top 50 most frequently mutations (middle panels); the base mutation type distribution of each sample (bottom panels). In the middle panels, genes mutation frequency and mutation type are indicated on the right. c Lollipop mutation diagrams of primary (pointing up) and metastases (pointing down). Different color patches represent different domains, and y-axes means the number of mutations in primary and metastases, note that multiple mutation in a gene may occur from some case (color figure online)
Moreover, the mutation information of the top 50 genes with the highest frequency, as well as the clinically important genes including KRAS, NRAS, BRAF, and PIK3CA were summarized and illuminated in Fig. 2b., to further illuminate the characteristics of mutated genes in each patient. The mutation frequency of PIK3CA was 43% (cases CC03, CC07, and CC08), two types of mutations were observed in which H1047R was dominant; whereas KRAS mutation was detected in only two patients (i.e., missense mutation in CC01 and multi-hit mutation in CC08). Notably, the mutation of either PIK3CA or KRAS in the primary lesion did not variate but was missing in metastatic foci (Table S1). Unexpectedly, no mutations of NRAS and BRAF were detected in all of the enrolled samples, which might attribute to the small patient sample size.
The type of alterations exhibited diversity between primary lesions and multiple metastases as evidence from the landscape. For instance, OBSCN mutations in case CC03, were multi-hit in primary but nonsense in metastases. The difference of tumor mutational burden (TMB) between samples is obvious in CC01, CC03, CC04, CC05 and CC06, the metastases had a higher TMB, but no significant difference was observed between primary and metastasis (Fig. S1a). Additionally, heterozygous HLA-I genotypes were detected in all of patients (30/30, 100%). Next, all immunologically relevant genes were extracted from the ImmPort. The mutations were picked out based on the immune-associated genes reflecting different statuses in the immune context of microenvironment (Fig. S2).
Finally, the mutation gene loci were diagramed for the top 10 most frequently mutated genes and the clinically important genes of KRAS and PIK3CA, which were depicted by lollipops (Fig. 2c and Fig. S1b). Subsequently, we found different mutation loci between the primary and metastases in NEB, OBSCN and B4GALNT4. These results indicate the intricate heterogeneity between primary and metastases, even when they shared the same mutation genes, the mutation form or loci varied greatly.
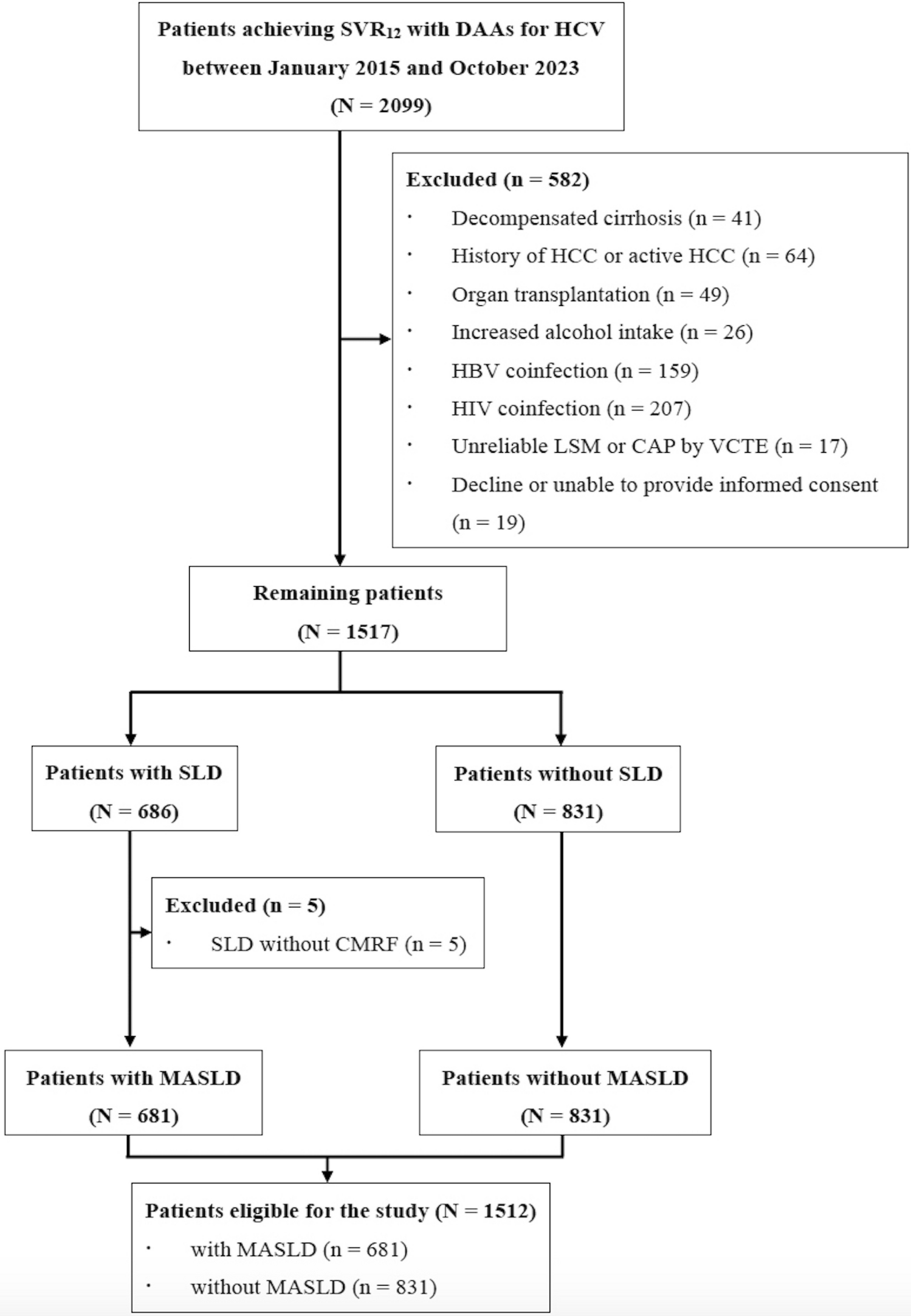
Intra- and inter- tumor heterogeneity in treatment-naive CCLMIntra-tumor and inter-tumor heterogeneity can act as a substrate for genetic diversity and tumor evolution. Pyclone, a software to predict subclone numbers and infer clonal structures, was employed to evaluate the heterogeneity of CCLM. Nine clusters were identified across all samples in case CC01, and different cluster included diverse mutations, for example, cluster 0 obtained 102 somatic mutations, and cluster 1 included another 81 mutation genes. The cellular prevalence displayed differently across the primary and matched multiple metastases, the variation trend was obvious in cluster 1 (mean 0.260–0.536), but seemed unchanged across samples in cluster 3 (mean 0.318–0.320) (Fig. 3a, b). Consistently, the similar phenomenon was also observed in the other seven patients (Fig. S3 a-b), indicating the intra- and inter-tumor heterogeneity.
Fig. 3
Intra and inter-tumor heterogeneity and subclones functional analysis. A The cellular prevalence of mutational clusters (vertical axis) in each sample is depicted at the left, width of the violin plots indicating the distribution of probabilities is generated by PyClone. The clusters order is along the x axis, and and n represents the amount of somatic single nucleotide variant included in that cluster. Composition profiles calculated from the PyClone model (right) plot the mean cellular prevalence of the variants in each cluster in each sample. Vertical lines at each point represent one standard deviation. b The Circos plot is performed by Metascape, it shows how genes from the clustered mutation gene lists overlap. On the outside, each arc represents the identity of each patient clustered mutation gene list. On the inside, each arc represent a gene list, where each gene has a spot on the arc. Dark orange color represents the genes that appear in multiple lists and light orange color represents genes that are unique to that gene list. Purple lines link the same gene that are shared by multiple gene lists. Blue lines link the different genes where they fall into the same ontology term (the term has to statistically significantly enriched and with size no larger than 100). c Enrichment analysis is performed by Metascape. The heatmap cells are colored by their p-values, light gray cells indicate the lack of enrichment for that term in the corresponding gene list (color figure online)
Next, a circus plot was applied to demonstrate the relationship between the identified mutation genes in all clusters. As shown in Fig. 3c, although patients shared fewer genes with each other (purple links), they exhibited more functional overlaps (blue links). This finding implied that mutations might be involved in different biological processes. In addition, we subsequently conducted gene set enrichment analysis of biological functions for the inferred clusters. Accordingly, two cohorts were hierarchically clustered based on Kappa-statistical similarities among their gene memberships (Fig. 3d). We hypothesized that these two cohorts may be linked to different prognoses. Further study involving more cases is required to confirm our findings.
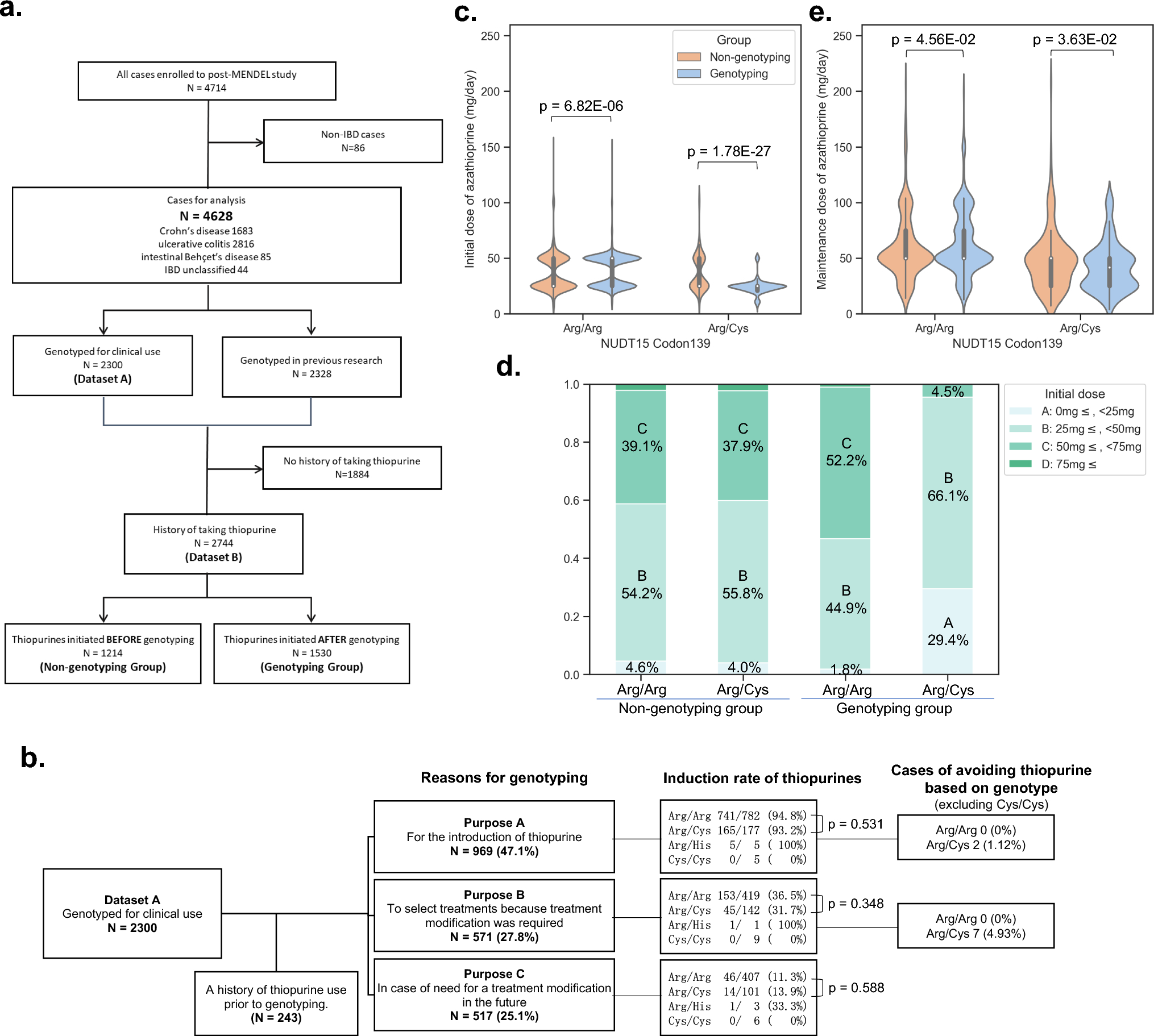
Three different phylogenetic evolution model of CCLMWe also sought to elucidate the evolutionary process of CCLM. Therefore, the Lineage Inference for Cancer Heterogeneity and Evolution (LICHeE) method was used to analyze the evolutionary model of eight patients. Three different phylogenetic models were identified (i.e., parallel, linear, and branching evolution) (Fig. 4). As shown in Fig. 4a, sibling clades deriving from a single ancestral genotype were found in two cases (i.e., CC04 and CC06), indicating parallel evolution in primary lesions and different metastases. For cases CC04 and CC06, there were no novel mutations detected in the liver metastases; all mutations evolved from the primary lesions. Moreover, a linear evolution model was noted in case CC03. Both liver metastases lost 36 mutations compared with the primary lesion, whereas one metastasis gained 45 novel mutations (Fig. 4b left). According to the LICHeE algorithm, we concluded that L1 first metastasized from the primary lesion, followed by L2 (Fig. 4b right).
Fig. 4
Tumor phylogenetic trees of colon cancer with multiple liver metastases. Phylogenetic trees inferred by LICHeE are shown for each patient (a, and left in b, c), showing each sample evolutionary process. The number in each node is the amount of somatic single nucleotide variant included in that subclone, the decomposition for each sample in the tree is displayed and how much each node contributes to the genomic makeup of the sample. Schematic outlines of tumor metastatic progression are shown on the right in B and C, with the clonal subpopulation of each sample shown. Arrows represent seeding clones between samples. Two possible scenarios are investigated in case CC05 and CC07, and four possibility in case CC08. GL, germ line. The asterisk stands for transition subclones
Another model revealed mixed lineages evolution in the rest cases, displaying diverse subclones in primary and metastases (Fig. 4c). Compared to primary sites, all the metastatic sites developed novel mutations to some extent, these mutations were formed during tumor progression, some of which acquired metastatic potential. For example, in case CC01, mutations were grouped into 5 subclones, in which subclone with 33 mutations was found in both primary and metastatic sites. During tumor progression, L2 and L3 first moved to liver after primary site developed subclone with 94 mutations, followed by developing shared 31 mutations respectively, while primary site developed other 36 mutations, and L1 transferred to liver.
Based on the same procedure, we speculated the evolutionary process from the phylogenetic trees in case CC02 (Fig. 4c). L2 was derived from L1 from subclone7, and evolved into a unique subclone 6 at the liver metastases, which is also consistent with a metastasis-seeding metastasis model. For cases CC05 and CC07, we investigated two possible scenarios according to the phylogenetic trees. For example, in case CC05, the L2 was derived from subclone 8, which could be acquired from the primary lesion or the L3 metastasis. There were four possibilities in case CC08: L1 and L3 could emerge from the primary lesion or the L2 metastasis in subclone 1.
Overall, these data indicated that the ancestral genotype may not be present in all samples. The proportions of subclones also exhibited diversity, and the dominant genotype was also distinguishable between samples. Thus, the evolutionary models of CCLM were complex and the subclones in primary lesions and multiple metastases were diverse, leading to tumor heterogeneity in a single patient.
The different functional effects of subclonesCancer is a disease of clonal evolution. The selected subclone might lead to cell proliferation, invasion, and metastasis. The subclone in a metastasis might possess the ability to induce different features compared with the genotype of the primary lesion. Hence, pathway and process enrichment analysis was carried out for six patient with different subclones in primary and metastatic lesions using Metascape (Fig. 5a) [23]. As illustrated in the heatmap, several enriched pathways were frequently observed in primary lesions and metastases from all eight patients. These included ATP-dependent activity, calcium ion binding, actin binding, and cell adhesion molecule binding; the latter was observed at almost all sites in these eight patients. In addition, immunohistochemically (IHC) analysis of cell proliferation and apoptosis was performed to further examine biological differences between primary and metastatic lesions in the same case (Fig. 5b, Table 2, Fig. S3, S4). According to IHC staining for cleaved-caspase-3, similar results were obtained for the primary lesions and metastases. However, in case CC03, the staining intensity for the primary lesion was weak and the positive area was 50%. For CC03.L1, the staining intensity was weak and the positive area was 80%. For CC03.L2, the staining intensity was moderate and the positive area was > 90%. Compared with CASP3, Ki-67 exhibit broader heterogeneity in cases CC01, CC05, CC08, CC02, CC03, and CC07.
Fig. 5
The functional analysis between the primary and metastatic lesions. (a) Gene Ontology (GO) was employed to enrich the mutated genes in the involved terms of molecular function, and the significant terms were hierarchically clustered based on Kappa-statistical similarities, among which those with the best p-value were selected and subjected to drawing a heatmap colored by their lg (p-values). Some terms, including ATP-dependent activity, calcium ion binding, actin binding and cell adhesion molecule binding, involved in almost all samples were emphasized in colorful. b IHC demonstrated the differential expression of active cleaved-caspase-3 and ki-67 between primary and paired metastatic lesions from CC01, which was observed in ki-67 staining but not in active cleaved-caspase-3 staining
Table 2 Expression of Caspase-3 and Ki-67 in primary and metastasis tissues by the immunofluorenscence staining
留言 (0)