
記住我
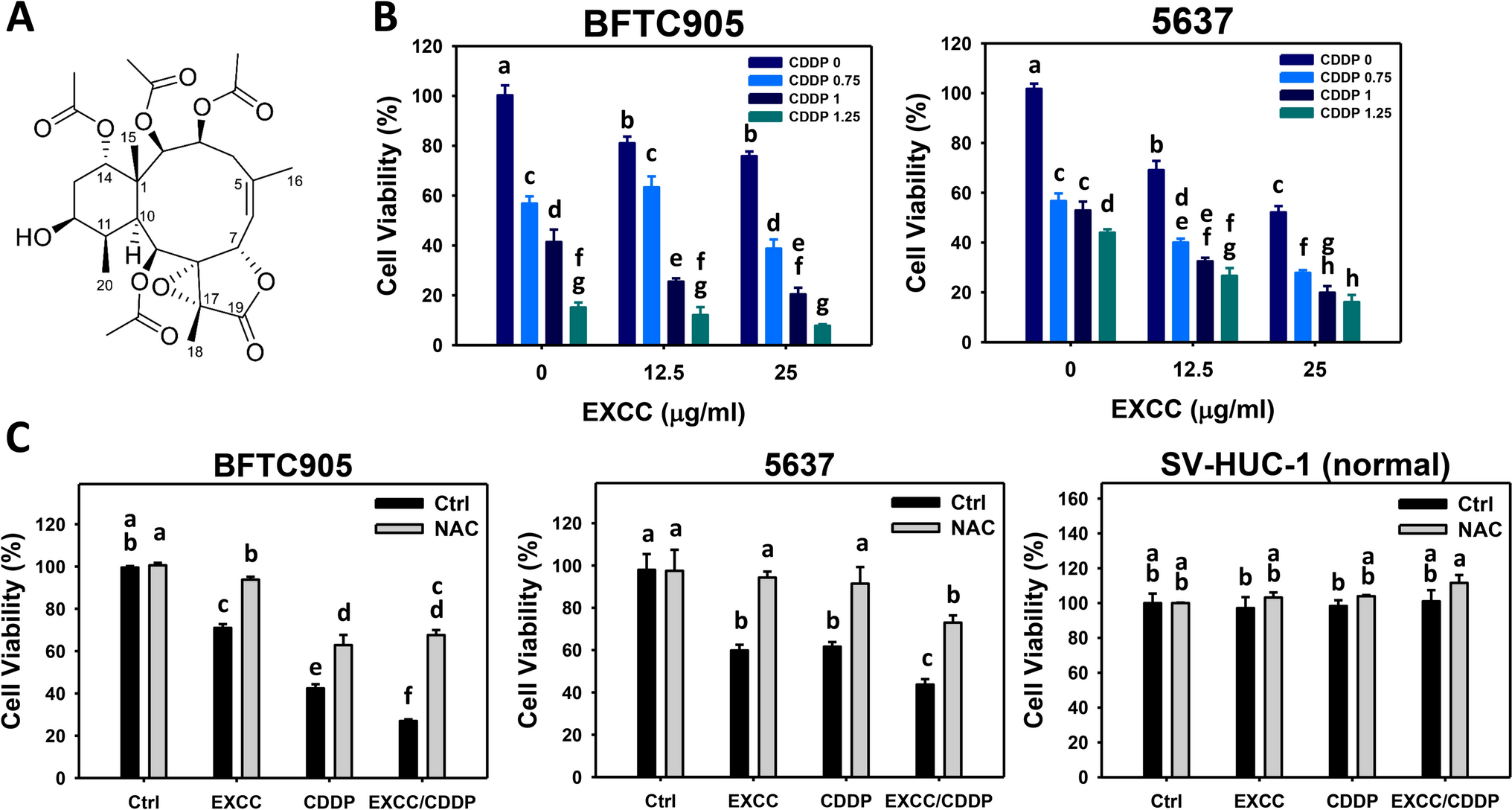
To improve the metabolite annotation, samples were analysed using two complementary methods. According to their distinct physicochemical properties, the chromatographic separation of highly polar metabolites was best achieved in the Synergi Hydro RP (Method 1); while metabolites with lower polarity showed a better chromatographic separation in the Waters Acquity column (Method 2).
The data set of metabolites identified from both chromatographic methods was combined. Data retrieved using method 1 and method 2 included 67 and 49 identified metabolites, respectively.
The workflow of the research undertaken, its rationale and the overall strategy applied is shown in Fig. 1. In the study, a training set of compounds containing drugs representative of the different mechanisms of hepatotoxicity were used. Upon cell incubation at two different concentrations, cell extracts were analysed by UPLC–MS to retrieve a broad range of signals that enabled the identification of metabolic patterns associated with specific mechanisms of drug-induced hepatotoxicity, different from those of general cytototoxicity.
Fig. 1
Workflow diagram of the research undertaken. Using a training set of compounds at IC10/IC50 concentrations, metabolomic data were retrieved by two analytical procedures and predictive models of toxicity (TOX) and hepatotoxicity mechanisms (OS, MI, APT, ST) were subsequently built. The biomarkers identified so far, for global toxicity and for each toxicity mechanism were discussed and examined in the context of metabolic pathway analysis. A test set of 87 compounds containing hepatotoxic compounds acting through different mechanisms, as well non-hepatotoxic compounds was subsequently evaluated and results were displayed in a radar chart, in order to account for the participation of the different mechanisms of toxicity and the outcome of this informtion discussed and compared to the occurrence of mechanisms previously reported in the literature
For model generation, we used the metabolome of cells treated with compounds predominantly acting via one mechanism of hepatotoxicity, and compared with the metabolome of cells treated with the rest of the compounds, starting at the lower concentration, and further comparing with the metabolome of cells treated with non-toxic compounds and non-treated control cells. At the IC50 concentration, only alive and attached cells metabolomes were analyzed and both types of signals, those related to general toxicity as well those linked to the occurence of a specific mechanism of hepatotoxicity were recorded.
Following this, biomarkers emerging as specific for global toxicity and for individual mechanisms of toxicity were properly identified. Metabolic pathway analysis was also conducted to confirm the relevance of each metabolite to the corresponding mechanism of toxicity. We then built predictive model based on these biomarkers relevant for global hepatotoxicity and for each of the individual mechanisms of hepatotoxicity. Following this, the developed models were further validated by examining the metabolomic pattern of toxicity of cells incubated with a larger set (87) of test compounds.
Global drug-induced hepatotoxicity modelA prediction model for assessing global hepatotoxicity (TOX) was constructed. Autoscaled metabolic profiles from cells exposed to two toxic concentrations IC10 and IC50 (Table 1) were selected and compared with profiles obtained from cells exposed to equivalent concentrations of non-hepatotoxic compounds, as well non-treated cells (controls). These two concentrations were selected to produce a broad range of signals for mechanism modelling. For the model generation, we used the metabolome of cells treated with compounds predominantly acting via one mechanism of toxicity, starting at the lower concentration (IC10), and compared with the metabolome of cells treated with non-toxic compounds and non-treated control cells. The IC50 concentration, as determind by the MTT test, is a concentration that affects 50% of the mitochondrial activity, while cell viability is much less affected. The metabolome obtained from cells treated at high concentrations (IC50) may contain signals attributable to general cytotoxicity events, overlapping, but not abolishing, the signals linked to a specific mechanism of drug-induced hepatotoxicity. Therefore, at this concentration we are recording both mechanism-related signals and general toxicity signals as well. Later, by doing these comparisons for each mechanism, at high and low concentration, we were able to distinguish the metabolites specifically altered by each toxicity mechanism from the metabolites arising in general toxicity, irrespective of the drug used. Hence, this strategy enabled us to discriminate relevant metabolomic changes linked to each mechanism of toxicity, from those less specific arising from a general cytotoxicity event (see below).
As a first approximation, we performed an unsupervised Principal Component Analysis (PCA) of the autoscaled data signals of identified metabolites from cells treated with drugs at IC10 and IC50. The four first principal components explained approximately 60% of the data variance. For better visualisation, six PCAs are shown in Fig. 2, showing the main (MI, OS) and secondary (APT, ST, CHOL) mechanisms of toxicity. In the PCA scores, only a slight separation of the samples according to the different mechanism of hepatotoxicity was observed when analysing the whole set of data. The analysis of the spatial distribution of objects in a PCA scores space is widely used to identify the main sources of variation and to get a first overview of the data structure and to identify potential outliers. However, this is an unsupervised approach and the interpretation of the sources of variation might be difficult, if they are the result of a combination of biological effects associated with one or more (orthogonal) principal components. Besides, other elements, like the technical and instrumental effects, might also have an impact (indeed, they can be one of the main sources of variance). Thus, even if in a PCA score plot there is no clear sample clustering associated with a given intervention (e.g. toxicity mechanism), it cannot be ruled out that there might be an impact on the metabolomic profile. Notwithstanding, what can be observed is that there is a clear separation trend among toxic and non-toxic concentrations/compounds.
Fig. 2
Principal component analysis on training set. PCA scores plots of the combined metabolome from cells treated with IC10 and IC50 concentrations of hepatotoxins acting through different mechanisms (oxidative stress (OS); mitochondrial disruption (MI); no toxic, (upper row); steatosis (ST); apoptosis (APT); cholestasis (CHOL), non toxic (NT), lower row; as well as with non-toxic xenobiotics, not assigned (NA) and controls. There is a clear separation trend among toxic and non-toxic concentrations/compounds
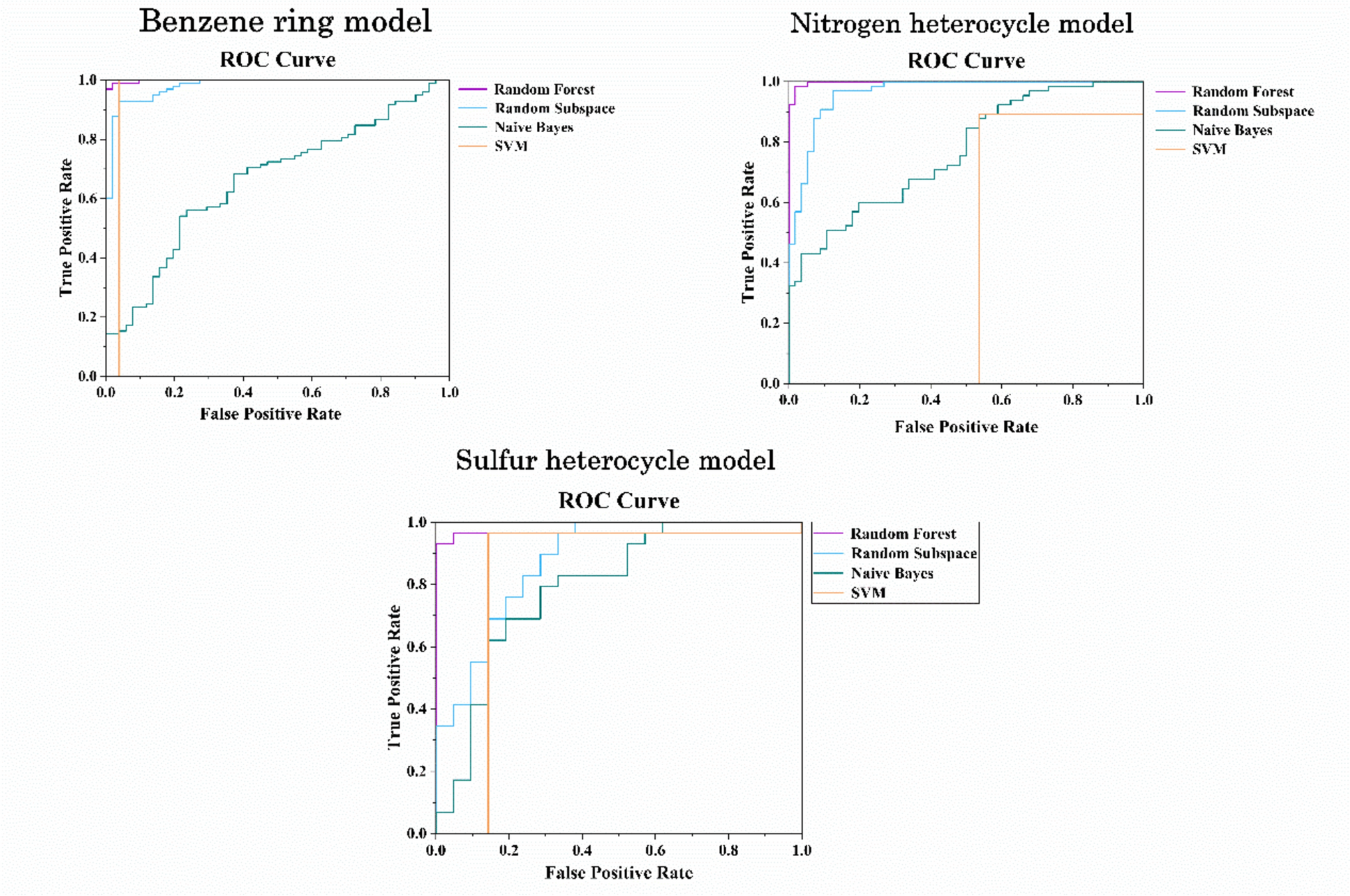
This first approach was followed by a partial least squares-discriminant analysis (PLS-DA) performed with data of the identified metabolites from cells incubated with toxic compounds at IC10 and IC50 vs non-toxic and control samples. In Table 2, we can observe the distribution of the samples in the different classes of the prediction models. We carried out a repeated (n = 10) random threefold CV (Lee et al. 2018) and 6 LVs were selected according to the lowest value of error rate (Supplementary Fig. 2a). Figure 3a shows the receiver operating characteristic curve (ROC) estimated by CV. The value of the area under curve (AUC) was 0.8, indicating a high performance for toxicity classification and a good sensitivity and specificity in discriminating hepatotoxic compounds from controls and non-toxic compounds. The statistical significance of the PLS-DA model was assessed by permutation testing (200 permutations, p value < 0.05) as described elsewhere (Neubert and Brunner 2007), using the AUROC as target function.
Table 2 Balance of positive and negative probes in the training set for each class and mode of action. Oxidative stress (OS); mitochondrial disruption (MI); steatosis (ST); apoptosis (APT); and cholestasis (CHOL)Fig. 3
ROC curves for global and individual toxicity mechanisms prediction models. The average ROC curves for TOX (a), OS (b), MI (c), APT (d) and ST (e), with standard deviation (std) on shadow, as well the AUC value (mean ± std). Top VIP scores for the corresponding prediction model (f–j). Red circles indicate higher metabolite concentrations in the corresponding toxic mechanism. On the contrary, blue circles refer to metabolites displaying high concentration in the negative mechanism group VIP scores of each model for all metabolites are displayed on Supplementary Table 2
Predictive models for the different mechanisms of drug-induced hepatotoxicityThen, a set of classification models to enable the discrimination among toxicity mechanisms (OS, MI, APT, ST and CHOL) was built using the metabolic profiles of cells incubated with the appropriate model compounds (Table 1). Accordingly, for each of the five considered mechanisms of toxicity a one vs all discriminant model was built, in which samples obtained from cells incubated with compounds inducing a given type of toxicity were compared to samples obtained from cells incubated with either non-toxic or compounds inducing a different type of toxicity.
We made a comparison of metabolomes from cells treated with low concentrations of toxic compounds at IC10 concentration, non-toxic compounds and non-treated cells. This was complemented with data obtained at higher concentrations (IC50) which enabled us to filter off signals attributable to general toxicity while emerging the mechanism-specific metabolome signals. In this way, the analysis for each mechanism allowed us to identify metabolites altered specifically by each toxicity mechanism and to exclude effects linked to general toxicity. Based on these metabolite changes, metabolic pathways analysis was also performed for each mechanism of toxicity and subsequently analysed and compared among them to confirm their relevance for a specific mechanism of toxicity.
Then, we built a predictive model based on the biomarkers relevant for global hepatotoxicity and those relevant for each of the individual mechanisms of hepatotoxicity (i.e., comparing the metabolome of cells incubated with compounds that exerted its action through a given mechanism with the rest of compounds). Model performance and statistical significance was carried out as for the Global drug-induced hepatotoxicity model. The number of LVs selected for each model is shown in Supplementary Fig. 2b–f. Supplementary Fig. 3 depicts the evolution of the sensitivity and selectivity of each PLS-DA model as a function of the discrimination threshold employed. Based on these results, the optimal classification thresholds for each model were 0.63, 0.35, 0.35, 0.28 and 0.19 for TOX, OS, MI, APT and ST, respectively (Supplementary Fig. 3). Figure 3a–e shows ROC curves for global toxicity model (a) and for each mechanism model (b–e) showing AUC mean values between 0.68 and 0.81, supporting the discrimination performance of the models. Nonetheless, the model built for the identification of CHOL toxic responses showed insufficient sensitivity and specificity (AUC < 0.5) (Supplementary Fig. 4). A possible explanation is that whereas HepG2 cells perform most of the mature hepatocyte metabolic functions, they have very limited ability for bile acid synthesis (Everson and Polokoff 1986) and have a reduced expression of the principal bile acids transporters (Kullak-Ublick et al. 1996), thereby limiting their applicability as an in vitro model for the detection of metabolic alterations caused by cholestatic drugs (Cooper et al. 1994).
A slightly lower predictive performance of the individual mechanistic models compared to the global hepatotoxicity model was observed. This could be attributed to the fact that there are no toxic compounds acting exclusively through a unique toxic mechanism, but to an interrelation of them within the cell which depends on the exposure conditions (i.e., concentration, time). Given this unavoidable overlapping effect of distinct mechanisms, it is understandable that the compounds, although classified as representative of one given mechanism of hepatotoxicity, may partially display biomarkers associated with other mechanisms of hepatotoxicity.
The most relevant metabolites for global hepatotoxicity assessment and for each mechanism prediction model (VIP > 1.5) are shown in Fig. 3f–j. Information about the identified metabolites is summarised in Supplementary Table 2 including m/z, retention time, HMDB code, metabolic pathway, VIP scores, and t test p value of each prediction model. Three relevant identified metabolites with VIP > 1 are grouped by type of mechanism and displayed in Fig. 4. Decreased levels of Glutathione (GSH) emerged as a consistent biomarker of oxidative stress mechanisms. GSH is involved in conjugation processes being of great relevance in liver detoxification processes (Kaplowitz 1981; Irie et al. 2016). Polyamines and their acetylated products N1-acetylspermine and N8-acetylspermidine showed high VIP scores in the MI, APT and ST models, and low VIP scores in the OS model. It is known that polyamines play an important role in the regulation of mitochondrial Ca+2 transport and ATP (Salvi and Toninello 2004) and are related to an increase of oxidative stress independent of the glutathione pathway (Rider et al. 2007). Moreover, the spermidine/spermine N (1)-acetyltransferase (SSAT) enzyme can be induced in the liver by toxins such as carbon tetrachloride (Matsui et al. 1981) resulting in an increase of N1-acetylated polyamines as well as SSAT is reported to be located mainly in mitochondria (Holst et al. 2008). Furthermore, in agreement with previous results ophthalmic acid was found highly significant (VIP > 1.5) in the OS and APT models. Besides, the concentration profile in OS is associated with GSH depletion observed in the course of oxidative stress (Soga et al. 2006). Ophtalmic acid resulted more relevant to OS, MI and ST, and differ from non toxic compounds.
Fig. 4
Boxplots of autoscaled peak intensities from a set of metabolites and their relevance in the mechanisms of hepatotoxicity. Toxic samples were grouped by mechanism as oxidative stress (OS), mitochondrial disruption (MI), Apoptosis (APT) and steatosis (ST). These biomarkers, typically absent in non-toxic compounds or controls, are equally recognised in the literature as biomarkers of the different types of hepatocyte damage. Autoscaling of each variable was carried out by mean centering followed by dividing by the standard deviation
Metabolic pathway analysisThe identification of the alterations in metabolic pathways associated with the different toxicity mechanisms was carried out by Metabolic Pathway Analysis (MetPA). For that purpose, MetPA was performed comparing samples from each mechanism versus control samples. MetPA combines several advanced pathway enrichment analysis procedures along with the analysis of pathway topological characteristics to help identify the most relevant metabolic pathways involved in a given metabolomic study (Xia and Wishart 2010). A list of all metabolites included and their corresponding HMDB ID are shown in Supplementary Table 2. Results from pathway analysis were summarised with 2 descriptors (− log10 (p value) and the impact factor) as described elsewhere (Xia and Wishart 2010), using metabolic pathways with > 3 hits for the analysis. Different metabolic pathways appeared significantly altered within each mechanism (Supplementary Fig. 5) when compared to controls. To determine whether these altered pathways were specific and could constitute a characteristic metabolic fingerprint of each mechanism of hepatotoxicity, a correlation analysis was performed to compare among the metabolic pathways altered in cells treated with drugs acting by one of the mechanisms with respect to controls cells, versus the metabolic pathways altered in cells treated with drugs characteristic of other mechanisms with respect to controls cells. The pairwise correlations between MetPA outcomes obtained for APT, ST, OS and MI were estimated using Mantel's test, as previously described (Ten-Doménech et al. 2021). In this study, the −log10(p value), and the impact factor (estimated as the sum of the importance of the measures of all metabolites in the pathway) were used as descriptors (i.e., coordinates) of the MetPA outcomes. Finally, the Euclidean distance between metabolic pathways was used as a measurement of dissimilarity between metabolic pathways. For a better understanding, this strategy is highlighted in the workflow shown in Supplementary Fig. 6. The correlation between dissimilarity vectors was estimated using the Pearson correlation as we previously described in detail (Moreno-Torres et al. 2021; Ten-Doménech et al. 2021). This test estimates a correlation score between the outcomes of two MetPA (ZM). A high, statistically significant score indicates a strong correlation between the two distance matrices containing the pairwise distances between the elements of each set, where small and large distances in one MetPA are associated with small and large distances in the second MetPA. Supplementary Table 2 shows the metabolites involved in each pathway. As described above, glutathione and other metabolites of the glutathione pathway were affected in the course of hepatotoxicity, being relevant to all mechanisms of toxicity. The nicotinate and nicotinamide pathway were altered only in the ST mechanism. Indeed, it has been already described in the literature that a decrease in the coenzyme NAD, characteristic of this pathway, is related to an increase in ST (Mukherjee et al. 2017) and we found this metabolite with VIP > 1 in the ST model. Results also agree with the literature in the sense that the glycerophospholipid pathway is altered in the course of MI and ST (Peng et al. 2018). Drugs inducing cholestasis did not significantly influence any metabolic pathway with the currently annotated metabolites. as previously mentioned. This is likely due to the fact that HepG2 cells lack the ability to synthesise, conjugate and transport bile acids (Everson and Polokoff 1986). Results depicted in Fig. 5 describe the correlation coefficient between paired comparisons of pathway analysis of the different mechanisms assessed and their statistical significance evaluated by the Mantel’s test. They show low, non-statistically significant correlations among the alterations observed for the different mechanisms. This result suggests that, despite some metabolic pathways are commonly altered across mechanisms, the overall impact on the metabolome remains characteristic of each toxicity mechanism. Thus, by evaluating the meta-analysis of results from the pathway analysis, a specific metabolic fingerprint for each toxicity mechanism could be obtained.
Fig. 5
Correlation between paired comparisons of pathway analysis evaluated using the Mantel’s test for the different mechanisms assessed. Changes in pathway analyses were inferred after comparing samples treated with drugs acting through a given mechanism of toxicity versus control samples. No significant correlation (permutation test p value < 0.05) was found among the results from metabolic pathway analysis observed for the different mechanisms. This indicates that impacts in the metabolic pathways are different for each mechanism of toxicity
Assessment of the global toxicity and toxicity mechanisms prediction models on a set of testing compoundsThe performance of global toxicity and toxicity mechanisms models to identify the contribution of each mechanism to the global hepatotoxicity were assessed in a test set of 87 compounds having been identified in the literature as acting via a principal mechanism of hepatotoxicity. Compounds were examined at a range of increasing concentrations (from 1 to 1000 μM), as an unknown compound would have been assayed in a blind in vitro test (Gómez-Lechón et al. 2010; Tolosa et al. 2012b; Chen et al. 2016), to see whether the model could reveal the occurrence of general toxicity and/or the contribution of the various mechanisms of toxicity at increasing concentrations (Concentrations assayed are reported in Supplementary Table 1; in some cases, the largest assayable concentration, because of compound's solubility is indicated).
PLS y-predicted values accounted for the magnitude of global toxicity, as well for the relative impact of each of the mechanisms of hepatotoxicity (OS, MI, APT and ST) associated with each compound at each assayed concentration. Low protein content in cell homogenates (less than 1/3 of that observed in blank samples) was a sample exclusion criterion, as it was indicative of extensive cell death that might distort the intracellular metabolome. Thus, these samples were excluded from the validation set (see the indication of low protein (LP)). Values for all compounds and all concentrations are summarised in Supplementary Table 3 together with the mechanism of toxicity attributed in the literature to each compound (Gómez-Lechón et al. 2010; Tolosa et al. 2012b). In Supplementary Fig. 7 the predicted toxicity index values of toxic compounds at the different concentration ranges (i.e., 0–1 µM; 1–10 µM; 10–100 µM, > 100 µM) is displayed. All compounds were examined at a wide range of concentrations, to assess whether the model identifies a general hepatotoxicity and/or the contribution of the various mechanisms of toxicity at each concentration assayed. Application of the model to each compound and each concentration revealed that a given mechanism of hepatotoxicity could predominate at a certain concentration, but at higher concentrations other mechanisms of toxicity would also be present and contribute to the compound’s global toxicity. Results showed, as expected, an upward trend in the toxicity index with increasing doses, irrespective of the toxicity mechanisms elicited by the drug.
As observed in the Supplementary Fig. 8, 80% of compounds named in the scientific literature as hepatotoxins were correctly classified as toxic when incubated at 1000 µM (or maximum concentration). It is also remarkable that the MI mechanism appears at lower concentrations as opposed to the other mechanisms where its incidence increases with concentration. MI is likely to appear in the early stages of toxicity mechanisms. Despite some disparity with the bibliography, each compound is classified within the mechanisms predicted in at least one of the concentrations used. It is remarkable to appreciate that our model predicts the occurrence of overlapping of mechanisms in more than 90% of the studied compounds. This seems to be the expected situation where two or more mechanisms occur at a time or sequentially.
Radar chart was used to integrate the outcomes from the analysis of the five models (TOX, OS, MI, APT, ST) and to visualise the participation of each of them in the validation compounds set (87), and to easily interpret the metabolic changes observed along the four concentrations tested. The participation of each mechanism of toxicity is represented in the radar chart; the different axes graphically represent the y-predicted values from each mechanistic model, in a relative scale, ranging from 0 (no participation) to 1 (full participation). Combined prediction plots of 8 representative hepatotoxic compounds, acting preferentially via a specific mechanism (Gómez-Lechón et al. 2010): (a) OS, (b) MI, (c) APT and (d) ST, are displayed in Supplementary Fig. 9. Results showed that although claimed in the literature to act preferentially via a given mechanism, such mechanism is not unique, and others are likely to be involved. Indeed, while the global toxicity index generally increased with concentration, the relative contribution of each toxicity mechanism to global hepatotoxicity was influenced by the concentration being assayed. Thus, although cumene hydroperoxide and Mercury II are claimed to cause OS (Gómez-Lechón et al. 2010; Tolosa et al. 2012b), and indeed this is the case according to metabolome analysis, other metabolic alterations (APT, ST and MI) are present at larger concentrations in cumene hydroperoxide, but not in Mercury II (Supplementary Fig. 9a). 2,4-Dinitrophenol and azathioprine (Supplementary Fig. 9b) are regarded as causing MI (Gómez-Lechón et al. 2010; Tolosa et al. 2012b). But this is more evident in 2,4 dinitrophenol at all concentrations, while in the case of azathioprine is more evident at lower concentrations, being overridden by OS, APT and ST at the highest concentration, in agreement with the bibliography. Aflatoxin B1 and etoposide are claimed as eliciting APT in hepatocytes (Gómez-Lechón et al. 2010; Tolosa et al. 2012b). This is indeed observed at higher concentrations in the case of aflatoxin B1, but it is less evident in the case of etoposide, where other mechanisms (OS) prevail over APT, although they are indubitably related (Supplementary Fig. 9c). Chlorpromazine and imipramine, claimed to cause ST (Gómez-Lechón et al. 2010; Tolosa et al. 2012b), do alter hepatocyte metabolome in this sense, at a high concentration, but other mechanisms are involved as well (Supplementary Fig. 9d).
To better illustrate the utility of the developed tools, we applied the models to three members of the same class of drugs, statins, potent hypocholesterolemic drugs, and compared their effects on the metabolome of HepG2 cells when assayed at 4 different concentrations (Fig. 6). Both atorvastatin and lovastatin showed, as mentioned in the literature, a mechanistic pattern of MI and OS, respectively (Gómez-Lechón et al. 2010). However, simvastatin, often described as apoptotic in the literature, shows a predominant OS pattern at the highest concentration. This apparent inconsistency can be explained, as suggested by other researchers, by the fact that the APT in simvastatin is preceded by an OS phenomenon (Qi et al. 2010).
Fig. 6
Integrative toxicity radar chart for 3 representative statins. The metabolome of cells incubated with three statins, described in the literature as causing MI (Atorvastatin), OS (Lovastatin) and APT (Simvastatin) as principal mechanisms of hepatotoxicity were examined at various concentrations. The global toxicity and the participation of the different mechanisms of hepatotoxicity were estimated accordingly with the models and represented using integrative radar charts. Contribution of the different mechanisms of hepatotoxicity at various concentrations is displayed. The participation of more than one mechanism is evidenced and a different degree of mechanistic contribution to toxicity is observed with increasing concentrations
In Supplementary Fig. 10, we have included 4 compounds that show remarkable changes in the mechanism of hepatotoxicity with concentration. Fluoxetine and Clozapine are named in the literature as causing OS; Troglitazone as MI and APT and Rifampicin as OS and MI (Gómez-Lechón et al. 2010; Tolosa et al. 2012b). In the case of Fluoxetine, the described mechanism of OS is correctly observed at concentrations of 100 and 1000 µM. On the contrary, Clozapine, also described as OS in the scientific literature, shows evolution from an MI mechanism at the lowest concentrations to an OS mechanism. Troglitazone at lower concentrations main mechanism was MI and is taken over by OS at higher concentrations as described in the bibliography (Smith 2003). Meanwhile, Rifampicin coincides with the literature description as causing MI at lower concentrations, while the mechanism that predominates at higher concentrations is OS (Chowdhury et al. 2006).
留言 (0)