Microarray data
Next-generation sequencing dataset (GSE135485) and methylation profiling dataset (GSE47359) were obtained from the GEO database. GSE135485 included 54 EMS samples and 4 normal endometrium tissue samples, based on GPL21290 Illumina Human HiSeq 3000 platform. GSE47359 consisted of 3 EMS samples and 6 normal endometrium tissue samples, based on the GPL8490 Illumina Human Methylation 27 platform.
On data processing and identification of differentially expressed genes (DEGs), R software (ver. 3.6.3, https://www.rproject.org/) were used to identify DEGs and differentially methylated genes(DMGs). The matrix file for GSE135485 was downloaded from https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE135485 and then gene IDs conversion was conducted with strawberry-Perl-5.30.0.1. The data normalization was done with the limma package and then processed with the edge R package to get DEGs. The cut off value of DEGs was set as |log2FC|> 4. P < 0.05 was considered to indicate a statistically significant difference.
Differential methylation genes (DMGs) identification
The HumanMethylation 27 BeadChip array, covers approximately 27,578 CpG sites at different gene regions, embodying the upstream region of the transcriptional start site, 5′untranslated region, exons, 3′untranslated region. The matrix file for GSE47359 was downloaded from http://ftp.ncbi.nlm.nih.gov/geo/series/GSE47nnn/GSE47359/matrix/.
The Champ package of R was used for the identification of CpG sites and DMGs with the threshold P < 0.05 and |log2FC|> 0.2. The Champ package is a highly integrated methylation analysis tool, matching the corresponding DMGs with the most differentially methylated CpG sites. A Venn diagram was used to illustrate the intersection between DEGs and DMGs. As a result, upregulated hypomethylated genes were listed.
GO term and KEGG pathway enrichment
Online analysis tool DAVID was used to conduct Gene ontology (GO) Enrichment Analysis of DEGs into the Cell Components(CC), Molecular Functions(MF), and Biological Processes(BP). All p values < 0.05 were considered to be statistically significant.
Patient recruitment
This study was initiated on November 11th, 2019 and terminated on April 20th, 2021. All of the women recruited in this study were being at child-bearing age and underwent laparoscopic surgery at the Department of Gynecology of Xiangyang Central Hospital. Five women with endometriosis were recruited before surgery. All these women had not received GnRH-a agonist or hormones treatment for at least six months and were preoperative diagnosed as an ovarian cyst. They were aged between 24 and 39 years old, mean ± SD (32.12 ± 4.90) years; Each case of endometriosis was staged during the operation according to the revised American Fertility Society classification of endometriosis (rAFS) and subsequently confirmed by histology. Among them, two were in rAFS staging III and the other three were in rAFS staging IV. All these patients were in the secretory phase of the menstrual cycle. Ectopic endometrium from the ovarian cyst of these 5 patients were obtained by laparoscopy.
Five women undergoing tubal ligation for sterilization were recruited as controls. All these five patients were aged between 28 and 40 years old, mean ± SD (34.60 ± 4.38) years. No minimal endometriosis was found in these control subjects and no hormones treatment for at least six months. All these women were in the secretory phase of the menstrual cycle. Normal endometrium were obtained by curettage during tubal ligation operation.
Cell culture
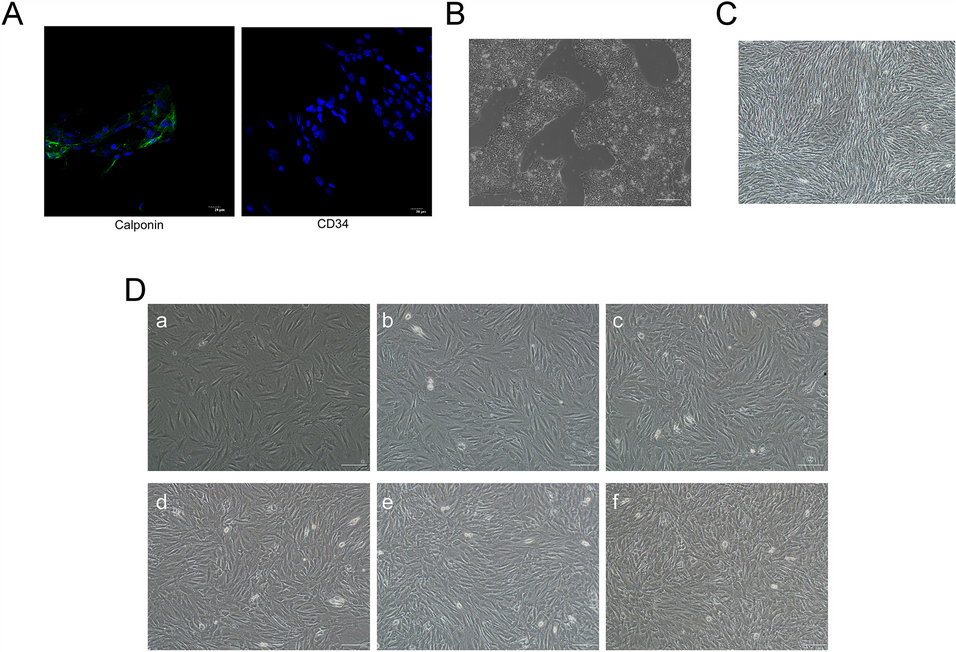
According to our previous study[36], tissues were washed with sterile Hank’s Balanced Salt Solution(HBSS, phenol-red-free) three times, then minced into pieces of approximately 1 mm3 and digested in 10 ml of HBSS containing 10 U/ml DNase I (Sigma) and type IV collagenase (0.03%; Sigma, St. Louis, MO) for 40 min at 37 °C. The supernatant was kept and epithelial cells and stromal cells in it were separated by differential centrifugation [21]. To repurify the endometrial cells, the selective attachment was carried out [22]. The endometrial cells were cultured in phenol-red-free DMEM/Ham’s F12 (Invitrogen, Carlsbad, CA) supplemented with 10% v/v fetal bovine serum (FBS; Invitrogen). Next, they were subjected to differential trypsinization and attachment for further purification. Finally, the primary epithelial cells were plated (2 × 104 cells/ml) in dishes in a culture medium as mentioned above. The detect the phenotypic characterization and ensure the purity of endometrial cell > 95%, the primary epithelial cells were tested by dyeing of vimentin and PCK.
Western blot
Western blot was performed according to our previous study [36] using primary anti-bodies against human SFRP2 (rabbit polyclonal, #HPA002652, Sigma-Aldrich, Merck, USA), anti-β-catenin (#ab6302, Abcam), DNMT1 (#ab13537, Abcam), and mouse monoclonal anti-β-actin (#A5441, Sigma-Aldrich) antibodies. The intensities of the protein bands were measured using the ImageJ (1.49 v) program.
5Aza-2′deoxycytidine (aza) treatment of EEECs
As deoxycytidine analogs, 5-Aza-CdR can be irreversibly mixed into DNA for synthesis, thus reducing the ability of DNA to accept methyl under the action of methyltransferase (DNMT). Meanwhile, 5-Aza-CdR forms a covalent complex with DNA methyltransferase (DNMT), reducing the activity of DNMT. And we want to decrease the methylation rate of the promoter of SFRP2 by using this drug. The EEECs were grown and treated with 1 μm of 5-Aza (Sigma-Aldrich #CAS 2353- 33-5) for 3 days for the inhibition of DNA methyltransferase activity.
Real-time RT PCR
Total RNA was isolated from EMS tissues and EEECs utilizing the TRIzol reagent (Invitrogen, Shanghai, China), and all cRNA transcripts were generated using a primeScript™ RT kit (Qiagen, Hilden, Journal of Molecular Histology1 Germany). All primers (Sangon Biotechnology, China) were listed as fellows: SFRP2, 5′-TGGGGGAAACGGTCGCACTC-3′, and 5′-GGCCACGAGACCATGAAGGAGG-3′. β-catenin, 5′-AAAGCGGCTGTTAGTCACTGG-3′ and 5′-CGAGTCATTGCATACTGTCCAT-3′. The qPCR was performed in triplicate to determine the relative levels of the target mRNA using SYBR premix Ex Taq™ Green II (Takara) in the CFX96 Touch sequence detection system (Bio-Rad, Hercules, CA, USA). Quantitative real-time PCR was conducted ABI 7500 Real-Time PCR System(Applied Biosystems/Life Tech).
Luciferase reporter assay
To detect the Wnt/β-catenin activation in EEECs, TOP/FLASH and FOP/FLASH reporter gene system (GenePharma Company, Shanghai) were selected to test the Wnt signaling pathway and the Promega dual-luciferase reporter gene assay system was used to measure the reporter activity. TOP/FOP values were used to represent the result. A higher value of TOP /FOP indicates a stronger Wnt pathway activity.
Methylation-specific PCR (MSP)
Genomic DNA from 5 ectopic endometrium and 5 normal endometrium was isolated using the DNA Extraction Kit (Sangon Biotech, Shanghai, China). In the 50ul system, DNA (2–5 µg) was denatured by NaOH (final concentration 0.2 mol/L) at 37℃ for 10 Min. Add 30 µL of 10 mmol /L hydroquinone and 40.5% sodium bisulfite to mix well, then incubate for 16 h in the condition of air isolation and out of light. The modified DNA passed by a DNA purification column and then eluted by water. At room temperature, it was modified with NaOH (the final concentration was 0.3 mol/L) for 5 min, and then precipitated with ethanol. Dissolve the DNA in 20µL water, stored at -20℃. Two pairs of specific primers were used to amplify the same nucleotide sequence of the tested gene using methylated primer pairs (M) 5′-GGAGTTTTTCGGAGTTGCGC-3′ and 5′-CTCTTCGCTAAATACGACTCG-3′, or unmethylated primer pairs (U) 5′-GTTGGAGTTTTTTGGAGTTGTGT-3′ and 5′-CTCTCTTCACTAAATACAACTCA-3′. The amplified products were detected by DNA agarose gel electrophoresis and analyzed by gel scanning.
Bisulfite sequencing PCR
Genomic DNA from 5 ectopic endometrium and 5 normal endometrium was isolated using the DNA Extraction Kit (Sangon Biotech, Shanghai, China). According to the manufacture’s instruction, and bisulfite modification was performed with the EZ DNA Methylation Gold Kit (Tianmo Technology, Beijing, China). Primer(Sangon Biotechnology, China) sequences for bisulfite sequencing were listed as follows: forward(M818-F)5′-TTTATGTTTGGTAATTTAGTAGAAATTT-3′ and reverse (M818-R) 5′-ATTTTACRTTAAAAATACCCCTCAC-3′. This area was 302-bp fragments including 28 CpG dinucleotides. The PCR conditions were: pre-denaturation at 95 °C for 3–5 min, denaturation at 94 °C for 30s, 55–60 °C for 30s, and 72 °C for 30s, 35cycles totally. Then, the sequence containing the SFRP2 sequence was sequenced(Sangon Biotech, Shanghai, China).
Plasmid construction and lentivirus production
The human SFRP2 lentiviral vectors were purchased from GenePharma and transfected EEECs according to standard manufacturer protocols. Furthermore, lentiviral vectors to knockdown DNMT1 expression were generated by the GenePharma Company, (Shanghai), and the interfering sequence is as follows: DNMT1-Homo-2664 GGAGCTGTTCTTGGTGGATGA. Three kinds of infection sequence were tested in the preliminary experiments, and one is useful as mentioned above. Post-infected cells were cultured for one week consecutively and lentivirus infection condition of target cells were determined by observing the expression time and intensity of GFP. To screen the stably transfection clusters, at the basis of transient infection, puromycin with minimum lethal concentration lasts for at least 4 days.
Immunohistochemistry
A cohort of 84 formalin-fixation paraffin-embedded specimens (FFPE), including 28 EMS ectopic endometrium, 28 eutopic endometrium and 28 normal endometrium were retrieved from Xiangyang Central Hospital from 2006 to 2020 with necessary clinical information. 28 eutopic endometrium and ectopic endometrium were get from 28 ovarian endometrial cyst patients which were aged between 25 and 43 years old, mean ± SD (35.05 ± 8.70) years; normal endometrium patients were aged between 29 and 48 years old, mean ± SD (41.80 ± 6.22) years. All the cases were reviewed by two senior pathologists separately again to ensure the diagnosis accuracy.
Immunohistochemical staining for SFRP2 was performed with 3-µm-thick sections using the Ventana Benchmark ULTRA automated staining system (Ventana Medical Systems, Tucson, AZ) according to the manufacturer’s protocol. SFRP2 (Abcam), the primary antibodies were added on the cell sections for two hours, Sections were incubated with a secondary antibody and visualized with 3, 3’-diaminobenzidine tetrahydro-chloride (DAB; Golden Bridge, Beijing, China). Sections were then subjected to nuclear counterstaining (blue staining) with hematoxylin. Two investigators were asked to review and score the anti-SFRP2 staining on the stained sections by adding the percentage score with the intensity score. Staining intensity was scored as 0 (negative), 1 (weak), 2 (moderate) and 3 (strong), while staining percentage was scored as 0 (< 10% staining), 1 (11–25% staining), 2 (25–75% staining) and 3 (≥ 75% staining). And these two fractions were added together, score 0–3: low; 4–6: moderate; 7–9: high.
Transwell assay
BD matrigel and 1640 were diluted in a ratio of 1:3 and 80ul was added to the upper chamber of the transwell chamber(8 μm; Millipore, Billerica, MA). EEECs were treated with 5-Aza, sh-DNMT1 or lentivirus carrying SFRP2-cDNA. Cell suspensions were configured according to the concentration of 200ul of serum-free medium containing 2.5 × 104 cells. 500 µl DMEM medium was added to the subchamber wells of the Transwell plate and the chamber was placed into the plate with care not to produce bubbles. Celcultures were grown in 37℃ incubator containing 5%CO2 for 24 h. Assays were then stopped by removing the non-invading cells in the top chamber with swabs. The chamber was removed and the medium was washed with PBS and the chamber was stained for 10 min; next the crystal violet of the cleaned chamber surface was washed with water, the cells in the upper chamber were wiped with a cotton swab and photographed under an inverted microscope. Cells in five visual fields per insert were counted (400× magnification).
Wound scratch assay
Log-growth EEECs were digested with trypsin and cells were evenly spread out into 6-well plates according to experimental grouping. They were incubated in an incubator at 5%CO2 and 37℃. When the cells grows to 80 -90% confluence, a straight line was drawn in the well using the appropriate pipette gun head along a sterilization ruler. The shed cells were washed out three times with PBS. In the presence of serum, untreated cells should migrate and fill the scratch area after approximately 48 h. Twenty-four hours after scratching, different treatments displayed remarkable effects on cellular migration in preliminary experiments, so this time point was chosen to end the assay. EEECs were treated with 5-Aza, sh-DNMT1 or lentivirus carrying SFRP2-cDNA as described above. Pictures were taken at 0 and 24 h under an inverted microscope. The relative migration length in five random fields was measured with ImageJ for further quantitative analysis.
Statistical analysis
All the experiments were repeated at least three times. SPSS 13 software was used for statistical analysis of all experimental data. The data were normally distributed. The comparison between the two groups was estimated by Student’s t-test. A p-value < 0.05 was considered significant. Chi-square was used in the Statistical analysis of immunohistochemistry data.



留言 (0)