
記住我



Agriculture needs to provide a significant increase in food, fuel, fiber, and fine chemicals in the next century to meet the needs of the growing world population. Different challenges exist in meeting these needs because of the effects of climate change, including an increased risk of drought and high temperatures, torrential rains, degradation of arable land, and the depletion of water resources (Atefi, et al., 2021). To mitigate these challenges, plant breeders are working to develop high-yielding, stress-tolerant crop varieties adapted to future climatic conditions and resistant to new pests and diseases (Fischer, 2009; Furbank and Tester, 2011; Rahaman et al., 2015).
Marker technology has been used in plant breeding since the 1980s, but it was not until 2001 when genomic selection (GS) was proposed by Meuwissen et al. (2001) to estimate all marker effects. Using this strategy, the full potential of markers was engaged, since GS is able to predict the output variable using all markers simultaneously in the model. The applications for GS continue growing, as it is employed by breeders in wheat (Triticum aestivum L.), maize (Zea mays L.), cassava (Manihot esculenta L.), rice (Oryza sativa L.), chickpea (Cicer arietinum L.), groundnut (Arachis hypogaea L.), etc. (Roorkiwal et al., 2016; Crossa et al., 2017; Wolfe et al., 2017; Huang et al., 2019). However, implementing GS in many plant breeding programs is challenging due to the many factors that affect its accuracy. Some of these factors include 1) genotyping quality, 2) not following the guidelines about where in the breeding program GS can be efficiently applied (Crossa et al., 2017; Yoosefzadeh-Najafabadi, et al., 2022), 3) insufficient number of lines in the reference (training) population, 4) appropriate allocation of samples and SNPs to training, testing, and validation using difference methods such as cross validation (Montesinos-López et al., 2022), 5) organization of field designs, 6) cross-validation strategy, tested lines in tested environments, tested lines in untested environments, etc., 7) heritability of the trait, 8) population structure, and 9) prediction model, etc.
There is empirical evidence that integrating high throughput phenotyping information collected by unmanned aerial systems (UASs), handheld scanners, tractor-mounted systems, and low orbiting satellite systems, in combination with genomic data, has the potential to complement GS and increase crop productivity. One of the advantages of recent phenotyping technology is that it can quickly and accurately obtain data on many agronomic traits (Atkinson et al., 2018). While the availability of new classes of phenomic information has fueled the development of a phenomic-based analog to GS, phenomic selection (PS), it is more probable that the integration of high throughput phenotypic information with other omics data is what can significantly improve the accuracy of GS. For example, Wu et al. (2022), using three omics datasets (transcriptomics, genomics, and metabolomics) as predictors, found that the integration of the three sources of information improves prediction accuracy in barley (Hordeum vulgare L.). Also, Hu et al. (2021) found that integrating multi-omics (transcriptomic, metabolomic, and genomics) data improved prediction accuracies of oat (Avena sativa L.) agronomic and seed nutritional traits in multi-environment trials and distantly related populations in addition to single-environment predictions.
Because phenotypic variation observed across diverse environments is a product of genetic and environmental variation, environmental information acts as a central bottleneck for the application of modern genomics-assisted prediction tools, especially for use across multiple environments. Thus, it is of paramount importance to incorporate high throughput environmental data into genomic prediction models to improve predictions in new environments with the same environmental characteristics (Rogers et al., 2021). Also, all environmental (historical and non-historical) data should be included as predictors to model the genotype by environment interaction more efficiently, which is key to increasing the prediction performance and genetic gain in breeding programs. The addition of environmental data in the modeling process is fundamental for more accurately predicting cultivars across diverse growing conditions (e.g., Jarquín et al., 2014; Messina et al., 2018; Millet et al., 2019).
As referenced, there continues to be a growing amount of empirical evidence that combining genomic, phenotypic, and environmental data is key to improving prediction accuracy (Montesinos-López et al., 2017; Cuevas et al., 2019; Krause et al., 2019). It is important to note that many robotics systems have been employed to measure plant orientation, plant height, leaf length, leaf area, leaf angle, leaf and stem width, and stalk count of many species such as sorghum (Sorghum bicolor L.), maize, cauliflower (Brassica oleracea L.), sunflower (Helianthus annuus L.), brussels sprouts (B. oleracea L.), and savoy cabbage (B. oleracea L.) (Jay et al., 2015; Fernandez et al., 2017; Baweja et al., 2018; Vázquez-Arellano et al., 2018; Vijayarangan et al., 2018; Bao et al., 2019; Breitzman et al., 2019; Qiu et al., 2019; Young et al., 2019; Zhang et al., 2020), architectural traits and density of the peanut canopy (Yuan et al., 2018), the number of cotton (Gossypium sp.) bolls (Xu et al., 2018), berry size and color of grapes (Vitis sp.) (Kicherer et al., 2015), and volume, shape, and yield estimation of vineyards (Lopes et al., 2016; Vidoni et al., 2017). Even the promising results of high throughput phenotyping face many technical challenges that need to be addressed regarding sensing, path planning, localization, obstacle avoidance, and object detection. More research is required to overcome these limitations of phenotyping robots and improve their accuracy, speed, and safety (Atefi, et al., 2021). Some publications that combine genomics and environmental information are Basnet et al. (2019), Monteverde et al. (2019), Washburn et al. (2021), Jarquin et al. (2021), Rogers and Holland (2022), Costa-Neto, et al. (2021a), Costa-Neto, et al. (2021b), among others. Few publications are available that integrate genomics, phenomics, and environmental information (Crossa et al., 2021).
In this study, using data from soft white winter wheat collected from 2019 to 2022 by Washington State University, we evaluated the prediction performance of integrating genomics and high throughput phenotypic information to predict grain yield under two scenarios of cross validation (CV), prediction of partially tested lines in tested environments using 7-fold cross validation (7FCV) and prediction of partially untested lines in untested environments using leave one environment out (LOEO) cross validation. These two strategies of CV were implemented using the Bayesian genomic best linear unbiased predictor (GBLUP) and the partial least squares (PLS) method.
Materials and methodsDatasets 1 to 4 (wheat data)Wheat lines used in this study are from the breeding program of Washington State University (WSU) and were grown at various locations in the state of Washington on grower-cooperator fields using common agricultural practices for the region. Grain yield (GY) was collected using a Zürn 150 Combine (Zürn Harvesting GmbH & Co.) and was used for each of the four data sets.
• Dataset 1, Wheat_1 (Year 2019) contains 1,397 unique lines and three environments (Kincaid, Lind, Pullman) and contains 1,869 total observations since some lines are repeated in various environments.
• Dataset 2, Wheat_2 (Year 2020) contains 758 unique lines and six environments (Farmington, Harrington, Kincaid, Lind, Ritzville, and Walla Walla) and contains 952 total observations since some lines are repeated in various environments.
• Dataset 3, Wheat_3 (Year 2021) contains 452 unique lines and eight environments (Davenport, Harrington, Kahlotus, Kincaid, Lind, Pullman, Ritzville and Walla Walla) and contains 780 total observations since some lines are repeated in various environments.
• Dataset 4, Wheat_4 (Year 2022) contains 363 unique lines and six environments (Davenport, Farmington, Harrington, Prescott, Pullman and Ritzville) and contains 483 total observations since some lines are repeated in various environments.
Phenotypic data was collected using the Sentera Quad Multispectral Sensor (Sentera, St Paul, MN), which covered target bands of interest for winter wheat evaluation. The camera has four sensors that cover eight broad spectral bands between 450 and 970 nm. An unmanned aircraft system (UAS) mounted with the Sentera camera flew a programmed route at an elevation of 45 m capturing overlapping georeferenced images. Collected UAS images were stitched and prepped for data extraction in Pix4Dmapper (Pix4D Inc., Denver, CO), creating a single orthomosaic image for each sensor per location. Orthomosaic images were transferred to Geographic Information System (QGIS) for plot identification and then further processed with a custom R code for calibration, index calculation, and single plot mean data extraction. In 2019, a single reflectance panel (85% reflectance) was used for radiometric calibration on red, blue, green, (RBG) and red edge bands (RE1 and RE2). Quantum efficiency coefficients were used to calculate near infrared (NIR) using:
NIR=2.921×Blue−0.754×RedThe NIR band was then normalized with a coefficient of 3.07 during the calculation of SRIs. In 2020 through 2022, a set of calibration panels were used (five panels ranging from 2%–85% reflectance, MosaicMill Oy, Vantaa, Finland). All raw band layers were adjusted based on the relationship:
Where the slope and intercept are based on the regression of the observed reflectance in calibration panels, digital numbers (DN) are the raw observed pixel values, and surface reflectance (SR) is the true reflectance value (Iqbal et al., 2018). All datasets used adjusted multispectral band values to calculate indices for further model analysis.
All the lines were genotyped using genotyping-by-sequencing (GBS; Poland et al., 2012). The original SNPs totaled 6,075,743, but after filtering for SNPs with homozygosity >80%, for less than 50% missing data, greater than a 0.05 minor allele frequency, and less than 5% heterozygosity, we end up with 19,645 SNPs. Markers with missing data were imputed using the “expectation-maximization” algorithm in the “R” package rrBLUP (Endelman, 2011). In each data set, the best linear unbiased estimates (BLUEs) were computed under two experimental designs:
For trials under an alpha lattice designThe BLUEs for GY within each environment were calculated using the lmer function of the lme4 package (Bates et al., 2015) of the R statistical software with the following mixed linear model:
yijkl=μ+gi+checki+tj+rkj+bljk+εijklwhere yijkl is the GY of the ith genotype in the jth trial, kth replicate and lth block, μ is the general mean, gi is the fixed effect of the genotype i, checki is the fixed effect of the check-genotype i, tj is the random effect of the trial, tj∼NIID0,σt2; where NIID stands for normal, independent and identically distributed, rkj is the random effect of the replicate within the trial, rkj∼NIID0,σr2; bljk is the random effect of the incomplete block within the trial and the replicate, bljk∼NIID0,σb2; and εijkl is the residual εijkl∼NIID0,σ2.
For trials under an augmented randomized complete block designIn this experimental design, the BLUEs for GY within each environment were calculated using the lmer function of the lme4 package of the R statistical software with the following mixed linear model:
where yij is the GY of the ith genotype in the jth block, μ is the general mean, gi is the fixed effect of the genotype i, checki is the fixed effect of the check-genotype i, bj is the random effect of the jth block, bj∼NIID0,σb2; and εij is the residual εij∼NIID0,σ2.
Bayesian genomic best linear unbiased predictor modelThe Bayesian models implemented only differ in the predictor they used. For this reason, the general model is given:
Where Yij denotes the response variable in the jth line in the ith environment, μ denotes the general mean (intercept), and ϵij are random error components assumed to be independent normal random variables with mean 0 and variance σ2. ETAijk is the predictor for the jth line in the ith environment. The different ETAijk´s (ETA) used are provided in Supplementary Table SA1.
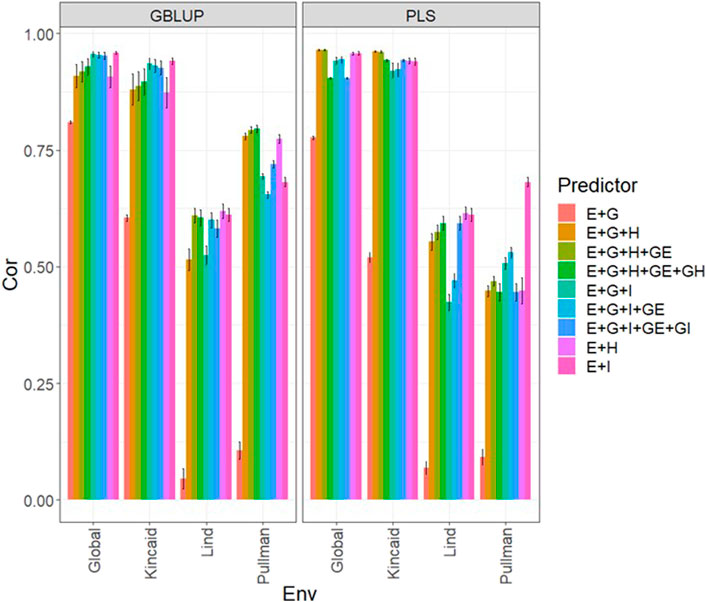
The vector of length eleven Hij1,…,Hij11 of the information include both the raw multispectral data and calculated indices denoted as: Blue, Green, Red, NIR, RE1, RE2, 900, 975 nm, NDRE1, NDVI, and Canopy Cover. While when the vector of length three (Iij1,Iij2,Iij3) was used as independent variables including only the indices: Can_Cover, NDRE1, and NDVI were used. The implementation of these models was carried out in the R statistical software (R Core Team, 2022) using the BGLR library of Pérez and de los Campos (2014). Equations used in the calculation of indices can be found in Table 1.
TABLE 1. Spectral reflectance indices implemented. The first column provides the name of the spectral indices, the second one its abbreviation, the third shows the equation used for computing each index, and the last one indicates the reference for each index.
Partial least squares modelThis study utilized the univariate PLS model, a statistical machine learning method introduced by Wold (2001) in econometrics and chemometrics for regression analysis. PLS is very useful for prediction problems where the number of independent variables (p) is larger than the number of observations (n) and when predictors are highly correlated. Under the univariate PLS framework, the response variable Y is a vector instead of a matrix of order n×1 that is linked to a set of explanatory variables (X) of order n×p (Wold, 2001; Boulesteix and Strimmer 2006). In PLS, instead of regressing Y on X, we regressed Y on T, where T are the latent variables (LVs), also called X-scores or latent vectors; these LVs are related to the original X and Y matrices. The goal of PLS regression is to maximize the covariance between Y and T; however, an iterative procedure is implemented for its computation. The main steps to compute the LVs under a univariate framework using the kernel algorithm for PLS are:
Step 1. Initialization of E = X and F = Y. Center each column of E and F; scaling is optional.
Step 2. Compute S=XTY (Cross product matrix) and then SST=XTYYTX and STS=YTXXTY.
Step 3. Compute the singular value decomposition (SVD) of SST and STS.
Step 4. Obtain w and q, the eigenvectors to the largest eigenvalue of SST and STS, respectively.
Step 5. Compute scores t and u as t=Xw=Ew and u=Yq=Fq.
Step 6. Normalize the t and u scores as t=t/tTt and u=u/uTu.
Step 7. Next, compute X and Y loadings as p=ETt and q=FTt.
Step 8. Deflate matrices E and F as En+1=En−tpT and Fn+1=Fn−tqT.
Step 9. Use as input En+1 and Fn+1, of Step 8, in Step 2, and repeat steps 2 to 9 until the deflated matrices are empty or the necessary number of components have been extracted.With the outputs of w, t, p and q vectors, the matrices W, T, P, and Q, respectively, are built. Finally, after having all the columns of W, we compute R as:
Next, with R we can compute the LVs, which are related to the original X matrix as:
Next, since we regressed Y on T, the resulting beta coefficients are b=TTT−1TTY. However, to convert these back to the realm of the original variables (X), we pre-multiplied with matrix R the beta coefficients (b); since T=XR,
To reach optimal performance of the PLS method, only the first a components are used. Since regression and dimension reduction are performed simultaneously, all B, T, W, P and Q are part of the output. Both X and Y are considered when calculating the LVs in T. Thereafter, predictions for new data (Xnew) should be done with:
where Tnew=XnewR. In this study, the optimal number of components was determined by cross-validation. We used the NRMSE, with an inner 10-fold cross-validation for selecting the optimal number of hyperparameters.In this application, we used the concatenation of the different sources of information for each predictor (ETA1 to ETA9 given in Table 1) as the matrix of independent variables X. For this reason, we first computed the design matrices of environments (XE), the design matrix of genotypes (Xg) and the design matrix of the Genotype × Environments term (XgE). But, since the PLS method does not allow direct inclusion, like the Bayesian GBLUP model, the genomic relationship matrix of lines G=MMTr, where M denotes the matrix of markers (coded as 0, 1 and 2) of order J×r; J denotes the number of lines; r the total number of markers. The design matrices of lines and genotype × environments were post-multiplied by their corresponding square root matrices of their corresponding relationship matrices to incorporate into the design matrix this relationship information. That is, instead of using only Xg; XgE as input, we used XgLg(with Lg=G0.5) and XgELgE (with LgE=GGE0.5). For this reason, the final input matrix used for ETA1 to ETA9 under the PLS model was; X=XE,XgLg;X=XE,XgLg,H;X=XE,XgLg,H,XgELgE; X=XE,XgLg,H,XgELgE,XgLg:H; X=XE,XgLg,I;X=XE,XgLg,I,XgELgE; X=XE,XgLg,I,XgELgE,XgLg:I; X=XE,H; X=XE,I respectively, where H=H11,…,H1J,…,HIJT; I=I11,…,I1J,…,IIJT; XgLg:H represents the interaction term between the genomic information and the multispectral information; XgLg:I represents the interaction term between the genomic information and three indices built from the multispectral information. We did not post-multiply the design matrix of environments (XE) since we did not compute an environmental relationship matrix with environmental covariates, only with the dummy values of the position of environments. For this reason, under the PLS model were used as input the vector of response variables (Y) and the input matrix X, just defined above. The implementation of the PLS models was performed with the R statistical software (R Core Team 2022) using the PLS library (Mevik and Wehrens, 2007).
Metrics for evaluation of prediction accuracyIn each of the four datasets (corresponding to years 2019–2022), for implementing the type of cross-validation partially tested lines in tested environments, we used seven-fold cross validation (7FCV) (Montesinos-López et al., 2022). For this reason, 7−1 folds (85.71% of the data) were assigned to the outer-training set and the remaining fold (14.29% of the data) was assigned to the outer-testing set, until each of the 7 folds were tested once. Under the PLS model for tuning the number of principal components required ten nested cross-validations, that is, the outer-training was divided into ten groups where nine were used for inner training set (90% of the training) and one for the validation (inner-testing) set (10% of the outer training). This means that under the PLS model, the data set was divided in outer-testing (14.29% of data), inner-training (77.14% of data), and validation (8.57% of data). Using the validation set, the optimal number of principal components was selected. Also, it is important to point out that the sum of the inner-training plus the validation equals the outer-training. Next, the average of the ten validation folds was reported as the metric of prediction accuracy to select the optimal hyperparameter (number of principal components). Then, using this optimal hyperparameter, the PLS model was refitted with the whole outer-training set (the 7−1 folds), and finally, the prediction of each outer-testing set was obtained. For the selection of the hyperparameters under the inner-training, the average mean square error was computed and used as the metric of accuracy, but for the outer-training to evaluate the prediction accuracy under the partially tested lines in the test environments cross-validation, the average Pearson´s correlation was computed. It is important to note that under the GBLUP, not tuning was required and only the outer 7FCV was implemented and the average of the 7 folds was reported as prediction accuracy for each environment using the Pearson´s correlation. But the computation of Pearson´s correlation across environments (Global) under the outer 7FCV was done between averages of true and predicted phenotypes of lines over environments per year (data set). On the other hand, to implement the cross-validation partially tested lines in untested environments we used a leave one environment out (LOEO) approach where the training set was composed of the total number of environments (nE ) minus one, and the remaining environment was used as testing set, meaning that each environment was used as testing set exactly one time. For this reason, only the average prediction accuracy for each environment was reported since only one-fold (testing set) was obtained for each environment. However, across environments, in addition to the average Pearson correlation, it was also possible to estimate the standard error. The Pearson´s correlation across environments (Global) in LOEO cross-validation was computed averaging the predictions resulting in each of the environments under study in each year. Under this approach the tuning process for the PLS was done exactly as was done under the 7FCV strategy.
ResultsThe results are provided in three sections. The first section provides, for each data set (each year), the variance component estimates of G, GE, residuals, and heritability. The second section outlines the results under tested lines in tested environments (7FCV) for each data set. The final section highlights the results under tested lines in untested environments for each data set. Supplementary Tables SA2, SA9 contain the results displayed in Figures 1–8.
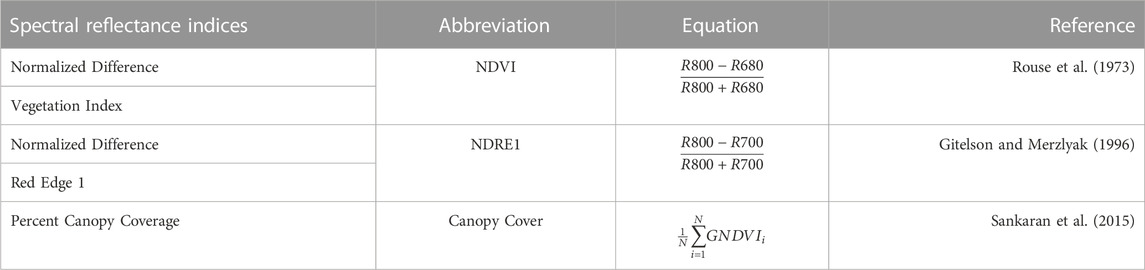
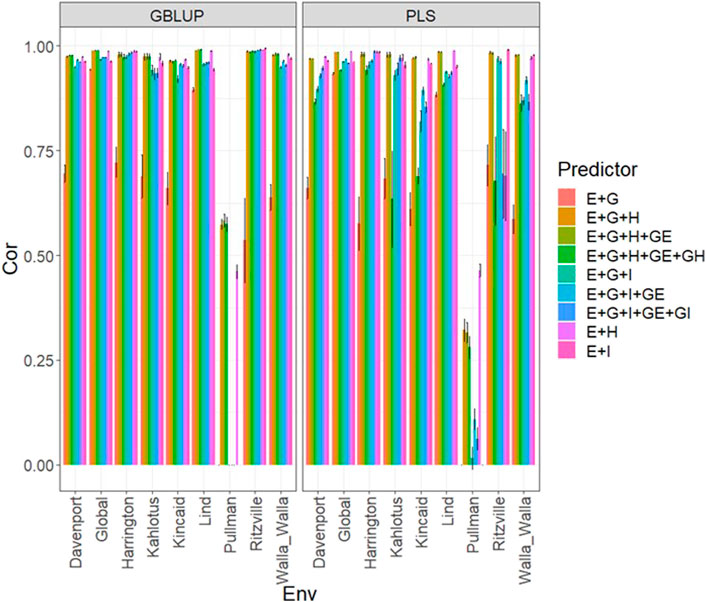
FIGURE 1. Dataset 1 (year 2019). Pearson´s correlation (Cor) and their corresponding Standard Error (SE) for each location and across location (Global) under tested lines in tested environments (7FCV) for nine evaluated predictors under a GBLUP and PLS models.
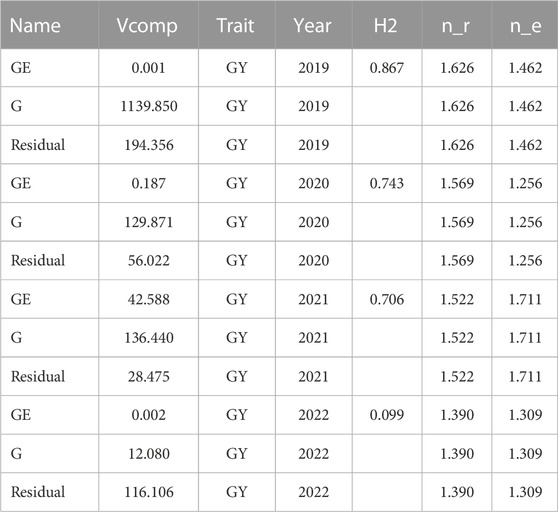
Variance components and heritabilityWe note in Table 2 that the heritabilities of years 2019, 2020 and 2021 are larger than 0.70; however, the heritability for year 2022 is low (0.099). Also, we can see in Table 2 that the GE interaction term is not relevant in years 2019, 2020 and 2022. We note that each of the four data sets is unbalanced since each wheat line was evaluated on average in 1.462, 1.252, 1.711, and 1.309 environments in 2019, 2020, 2021 and 2022, respectively. It is important to note that the environments evaluated in each year were 3, 6, 8, and 6, respectively. It can also be observed in Table 2 that the replications of each line in each environment were less than two since some lines were evaluated in replicated experiments while the remainder were examined in unreplicated experiments.
TABLE 2. Heritability estimates (H2) of grain yield (GY) in each of the 4 years and variance components (Vcomp) for the genotypes (G), genotype by environment interaction (GE) and Residual, n_e denotes the average number of locations, n_r denotes the average number of replications of each genotype. Year is the column to differentiate each data set.
Partially tested lines in tested environmentsThe results under 7FCV for each year are provided. It is important to note that for each model (GBLUP and PLS), nine predictors were evaluated to see how much each part contributes to improve the prediction accuracy and the results are reported for each environment and across environments (Global) for each year.
Data set 1 (year 2019)In Figure 1 and Supplementary Table SA2 we observe that under the GBLUP model, the best prediction performance was observed in environment Kincaid and the worst in Lind, while under the PLS model, the best predictions were also observed in the Kincaid environment, and the worst in Pullman. In Figure 1 and Supplementary Table SA2, the worst predictions were observed under the predictor (E + g). We observed when both types of information are integrated (genomic + multispectral information under its H or I versions) the best prediction performances were obtained and the predictions across environments and for some environments with Pearson’s correlation values close to one. However, Figure 1 shows that adding the interaction terms gE, gH, and gI to the predictors does not significantly increase prediction performance. Also, it is observed that simple predictors that contain only multispectral information, like E + H and E + I, produce similar performance to predictors that incorporate the genotypic information and interaction terms. However, the predictors that integrate both sources of information were more stable and consistent. Regarding the predictions using H or I, we observe that using I information is better since more consistent results than the H information and larger Pearson’s correlation are observed. In the models, we observe that both models are effective in this prediction problem and with this data, yet the predictions using GBLUP were better in most cases (Supplementary Table SA2).
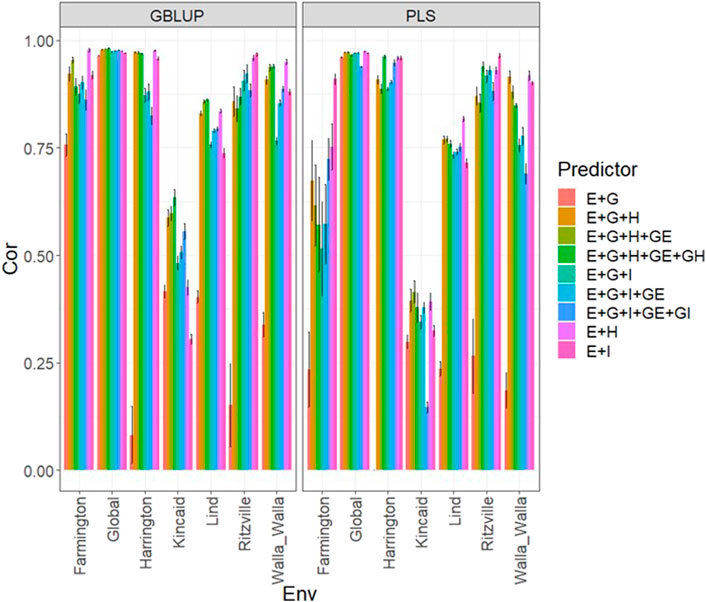
Data set 2 (year 2020)Under the GBLUP and PLS models, the best predictions were observed in environment Ritzville and the worst in Kincaid; however, very competitive predictions were observed in most environments except for Kincaid (Figure 2; Supplementary Table SA3). Also, the worst predictions were observed under the predictor E + g (see Figure 2; Supplementary Table SA3). The best prediction performances were observed when both types of information are integrated (genomic + multispectral l information under its H or I versions) with Pearson’s correlation values close to one in some environments and across environments (Figure 2). Again, we observe in Figure 2; Supplementary Table SA3 that adding interaction terms gE, gH, and gI in the predictors does not provide a relevant increase in performance. In the 2020 data, we observed simple predictors with only multispectral information like E + H and E + I produce a similar performance to more complex predictors that incorporate the genotypic information and some interaction terms, but predictors with both sources of information are more stable and consistent. Regarding the predictions using E + H and E + I, we observed the predictor E + H produced results closer to one for Pearson’s correlation values. Both models were effective in this prediction scenario and with this data, but the predictions using GBLUP were better (Supplementary Table SA3).
FIGURE 2. Dataset 2 (year 2020). Pearson´s correlation (Cor) and their corresponding Standard Error (SE) for each location and across location (Global) under tested lines in tested environments (7FCV) for nine evaluated predictors under a GBLUP and PLS models.
Data set 3 (year 2021)In Figure 3 and Supplementary Table SA4, we can note that under both models (GBLUP and PLS), the worst predictions were observed in Pullman. In the remaining environments, the predictions were effective since they were close to one in terms of Pearson’s correlation. The worst predictions were observed under the predictor E + g and the best when both types of information were integrated (genomic + multispectral information under its H or I versions) with Pearson’s correlation values also close to one. Again, it was observed in Figure 3; Supplementary Table SA4 that adding interaction terms gE, gH, and gI in the predictors did not improve the prediction performance. It was observed that simple predictors with only multispectral information like E + H and E + I, provided similar accuracies to predictors with both types of information (genotypic + multispectral information) and interaction terms. However, we observed more stable and consistent predictions in predictors that integrated both sources of information. The predictions using H or I did not produce relevant differences since, in some cases, using I information provided slightly better results than using H or vice versa. In the models, we observed both models were effective in this prediction scenario, and with this data, the predictions using GBLUP were slightly better (Supplementary Table SA4).
FIGURE 3. Dataset 3 (year 2021). Pearson´s correlation (Cor) and their corresponding Standard Error (SE) for each location and across location (Global) under tested lines in tested environments (7FCV) for nine evaluated predictors under a GBLUP and PLS models.
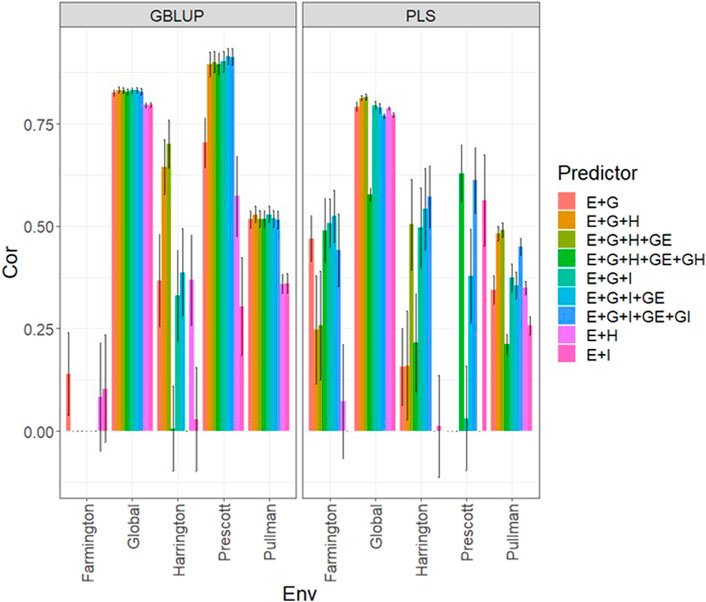
Data set 4 (year 2022)In Figure 4 and Supplementary Table SA5, we note that in the GBLUP model, the best and worst predictions were observed in Prescott and Farmington, respectively, while under the PLS model, the environment’s performance showed no difference. However, in both models, the predictions were not as high as those observed in the previous years. Also, the worst predictions were observed with the predictor E + g (see Figure 4; Supplementary Table SA5) and the best joining of the two types of information (genomic + multispectral information under its H or I versions). Again, we observed that adding interaction terms gE, gH, and gI, in the predictors did not improve the prediction performance regarding the additive integration of both types of information. Also, we observed that simple predictors with only multispectral information, like E + H and E + I, provided more competitive accuracies than predictors with both types of information (genotypic + multispectral information) and interaction terms. Yet we did not observe using the predictor E + H to give the best predictions. More stable and consistent predictions were observed in predictors that integrated both sources of information. Also, regarding the predictions using H or I, we observed non-relevant differences between them since sometimes using I information provides slightly better results than using H or vice versa. We observed that both models faced difficulties in predicting some environments but that the GBLUP model outperformed the PLS model (Supplementary Table SA5).
FIGURE 4. Dataset 4 (year 2022). Pearson´s correlation (Cor) and their corresponding Standard Error (SE) for each location and across location (Global) under tested lines in tested environments (7FCV) for nine evaluated predictors under a GBLUP and PLS models.
Partially tested lines in untested environmentsIn this section, the results under LOEO for each year are reported.
留言 (0)