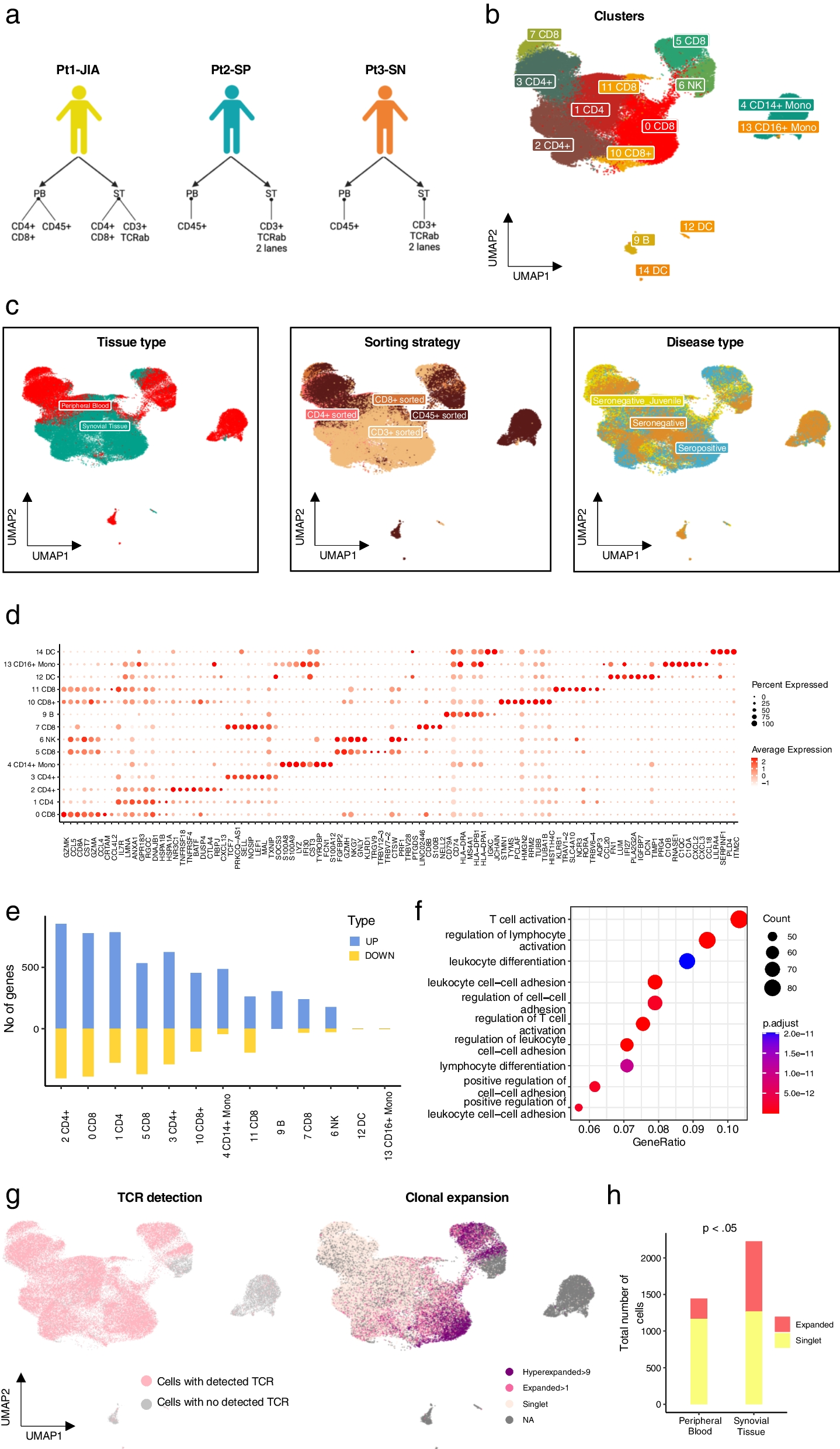
Analysis of GSE datasets
Expression profiles of skeletal muscles from healthy controls and CLI patients were obtained from the Gene Expression Omnibus dataset GSE 120642. The screening criteria for differential gene expression analysis was |log2 fold change (log2FC)|> 2, P < 0.05. The list of human kinases used for the analysis was taken from Zhang et al. (2021) and the KinHub List of Human Kinases. The tree file of the human kinome was courtesy of Cell Signaling Technology and was presented using the web-based tool KinMap (Eid et al. 2017). The expression changes and log2FC were reflected by the colors and size of the squares. Gene expression correlation in muscle between VEGFA and differentially expressed kinase was performed using GEPIA datasets.
Animal studies
All in vivo experiments were involved of 10 weeks male C57BL/6 mice (Liaoning Changsheng Biotechnology Co. Ltd., Liaoning, China), and approved by the First Affiliated Hospital of Zhengzhou University. Mice were housed in a 12-/12-h light/dark cycle with free access to standard mouse chow and water and acclimated to the facility 7 days before the experiment.
Experimental modelsIn vivo Matrigel plug assay
Matrigel plug assay was conducted according to the previously published methods (Meng et al. 2018). Briefly, adenoviral vectors for AURKA overexpression (Ad-AURKAOE) and its negative control (Ad-AURKAOE) were constructed, propagated, and titered. Matrigel mixed with 50 ng/mL VEGF and 30 U/mL heparin was injected subcutaneously into mice together with Ad-AURKAOE or Ad-NCOE. After a period of one week, the Matrigel plug was taken out for subsequent experimental manipulations. Hematoxylin and eosin staining was conducted to visualize the formation and infiltration of functional microvessels.
A diabetic mouse model with CLI
C57BL/6 mice were intraperitoneally administered with freshly prepared streptozotocin (STZ) solution (Shanghai yuanye Bio-Technology Co., Ltd., Shanghai, China, CAS. S17049) solution at a dose of 50 mg/kg body weight for five consecutive days to induce diabetes. Seven days after the first STZ administration, blood glucose levels were tested and only those mice with glucose levels above 15 mmol/L were included in the protocol. After the confirmation of diabetic mice, unilateral hind limb ischemia was surgically induced by femoral-saphenous artery-vein pair resection (Sarkar et al. 2009). For the gene transduction experiment, mice were anesthetized to induce hind limb ischemia using a procedure consisting of the ligation of both the proximal end of the femoral artery and the distal portion of the saphenous artery. Immediately, Ad-AURKAOE or Ad-NCOE (109 plaque-forming unit) was delivered to the ischemic adductor muscle and gastrocnemius muscles (Shaikh et al. 2017). The blood flow of the ischemic and the contralateral feet was sequentially measured (at 0, 3, 7, 14, 21, and 28 days post-surgery) by laser Doppler Image, and the relative perfusion ratio between the ischemic foot and non-ischemic foot was calculated. The behavioral motor deficit was assessed using the Tarlov-motor scoring system (Westvik et al. 2009) at the above time points. At 21 days post-ischemia, gastrocnemius muscles from terminally anesthetized mice were frozen or paraformaldehyde fixed for further analysis.
Cell cultures and treatment
Human microvascular endothelial cells (HMEC-1) (iCell Bioscience Inc., Shanghai, China) were cultured in EGM-2MV medium (Lonza, Basel, Switzerland) supplemented with fetal bovine serum (FBS) and growth factors at 37 °C in the presence of 5% CO2 in the air. To mimic diabetes in vitro, HMEC-1 cells were subjected to endothelial basal medium (EBM-2, Lonza) containing 25 mmol/L d-glucose (Aladdin, Shanghai, China) (high glucose, HG). To simulate ischemia-induced tissue starvation, cells were cultured in medium supplemented with no additional growth factors (nutrient deprivation, ND) (Caporali et al. 2011). Cells incubated with d-Mannitol (normal glucose, NG) and EGM-2MV medium supplemented with FBS and growth factors served as controls.
Prior to cell seeding, cells were infected with prepared Ad-AURKAOE or Ad-NCOE at a multiplicity of infection of 10 for 48 h. Cells were then detached and re-seeded on plates for subsequent analyses under different stimuli as described above.
Cell permeabilization assay
Cell suspension (200 μL) with a density of 5 × 104 cells/mL was added to the upper compartment of the Transwell chambers, and the lower compartment was filled with culture medium. After the establishment of confluent monolayers of cells, the medium was discarded and the washing procedures were repeated twice using PBS. A total of 100 μL fluorescein isothiocyanate (FITC)-labeled dextran (FITC-dextran, 1 mg/mL, Maokang Biotechnology, Shanghai, China) was added to the upper compartment, and 500 μL phosphate buffer saline (PBS) solution was added to the lower compartment. The cell permeability was assessed by measuring the fluorescence intensity of FITC-dextran from the upper compartment to the lower compartment.
Hematoxylin and eosin (HE) staining
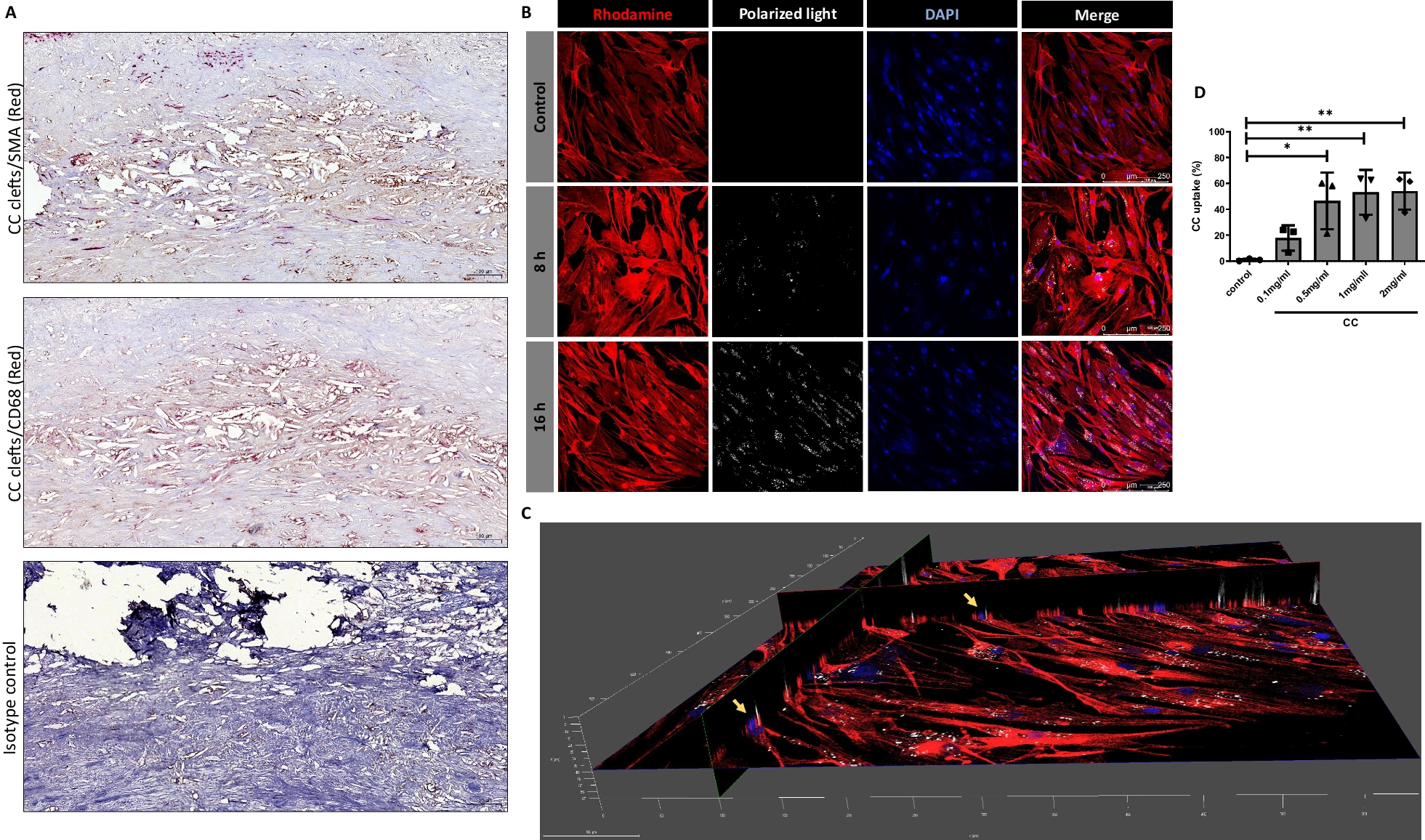
HE staining was performed accordingly to a standard procedure on 5 μm thick Matrigel plug sections and gastrocnemius muscles. Slides were stained with hematoxylin (Solarbio, Beijing, China) and eosin (Sangon, Shanghai, China), according to standard procedure. After the staining was completed, the number of capillaries of gastrocnemius muscle sections was quantified. The recognition and quantification was referred by Zaccagnini et al. (2019).
Immunofluorescent (IF) staining
HMEC-1 cells were fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton-X-100. The proliferation was visualized using bromodeoxyuridine (BrdU) staining. Slides were stained with anti-BrdU (1:100, ABclonal, Wuhan, China, #A20304), followed by Cy3-conjugated anti-rabbit antibody (1:200, Invitrogen, Carlsbad, CA, USA, #A27039). CD31 staining was performed on 5 μm thick Matrigel plug slides. Slides were stained with anti-CD31 (1:100, Santa Cruz Biotechnology, Santa Cruz, CA, USA, #Sc-376764), followed by Cy3-conjugated anti-mouse antibody (1:200, Invitrogen, #A21424). For functional vessel identification, mice were injected with FITC-labeled isolectin-B4 via tail vein, 30 min before sacrifice. Double IF staining was conducted on 5 μm thick gastrocnemius muscles. The perfused capillaries were labeled with anti-CD31 (1:100, Santa, #Sc-376764), followed by Cy3-conjugated anti-mouse antibody (1:200, Invitrogen, #A21424) and FITC-conjugated anti-rabbit antibody (1:200, Abcam, MA, USA, #AB6717). The density of small arteries (CD31+/α-SMA+) and capillaries (CD31+/α-SMA−) were double stained with anti-CD31 (1:100, Santa, #Sc-376764) and anti-α-SMA (1:100, ABclonal, A17910), followed by Cy3-conjugated anti-mouse antibody (1:200, Invitrogen, #A21424) and FITC-conjugated anti-rabbit antibody (1:200, Abcam, #AB6717).
Immunohistochemical (IHC) staining
IHC staining was performed accordingly to a standard procedure on 5 μm thick gastrocnemius muscles. Slides were stained with Desmin (1:100, ABclonal, #A3736) or 4-HNE (1:100, Abcam, #AB48506), followed by HRP conjugated anti-mouse antibody (1:500, ThermoFisher, Pittsburgh, PA, USA, #31430) or anti-rabbit antibody (1:500, ThermoFisher, #31460).
Cell proliferation and tube formation assay
Cell suspension with a density of 5 × 104 cells/mL was seeded into 96-well plates and cultured respectively for 12, 24, and 48 h. After incubation with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, KeyGEN, Nanjing, China) for 4 h at the indicated time points, the absorbance of each well at 490 nm was recorded to represent cell viability.
Cells (100 μL, 2 × 105 cells/mL), after indicated treatments, were seeded on plates coated with Matrigel. Tube formation was observed at 8 h under light microscopy (Olympus, Tokyo, Japan). The tube formation ability was evaluated in terms of the length and bifurcation points of the tube structure.
Scratch-wound assays
Cells grown in six-well plates were treated as per experimental requirements and cultured with 1 μg/mL Mitomycin C (Sigma-Aldrich, St. Louis, MO, USA) for 1 h prior to the scratch procedure. A liner scratch was created in the confluent cell monolayer using a sterile 200 μL pipette tip. Representative images were obtained at 24 h under a microscopy, and the migration capacity was defined as the blank distance at 0 h and at 24 h.
ROS and lipid ROS detection assay
Cultured cells were digested with 0.25% Trypsin–EDTA and centrifuged at 140g for 5 min, and then the cell precipitate was washed and re-harvested. 2′,7′-dichlorofluorescein diacetate (DCFH-DA, KeyGEN), diluted as 1:1000 in serum-free medium. Cells were pre-incubated with appropriate volume DCFH-DA solution for 30 min in the dark. DCFH-DA that failed to enter cells was removed with PBS. Cellular ROS was fluorometrically monitored by flow cytometry. The detection of lipid ROS was carried out in the same manner described above, except that the fluorescent probe C11-BODIPY (581/591) (Gibco, Grand Island, NY, USA) was used. ROS level in tissues were observed using a ROS staining kit (BestBio, Shanghai, China) following the manufacturer's protocol.
Immunoblotting
Tissues or cells were solubilized in RIPA lysis buffer (Beyotime, Shanghai, China) containing protease inhibitors (Beyotime), and the obtained lysates were prepared by centrifugation at 10,000g for 3 min at 4 °C. The protein concentration of the lysates was quantified using a Bicinchoninic acid assay kit (Beyotime). Equal amounts of denatured- protein samples were separated on SDS–polyacrylamide gel electrophoresis (PAGE) (Beyotime) and transferred on polyvinylidene difluoride (PVDF) membranes (ThermoFisher). The membranes were incubated with blocking buffer (5% [m/v] bovine serum albumin (BSA) in Tris-buffered Saline-Tween 20), following primary and secondary horseradish peroxidase-conjugated antibodies incubation. All antibodies were diluted in blocking buffer: anti-AURKA (1:500, ABclonal, #A2121), anti-VEGFA (1:1000, ABclonal, #A5708), anti-GPX4 (1:1000, ABclonal, #A11243), anti-SLC7A11 (1:1000, ABclonal, #A13685), anti-ACSL4 (1:1000, ABclonal, A16848), anti-ALOX5 (1:1000, ABclonal, A2877), anti-Cyclin B1 (1:500, ABclonal, A2056), anti-CDK1 (1:500, ABclonal, A0220), anti-p-PI3K (1:1000, ABclonal, AP0427), anti-PI3K (1:1000, ABclonal, A4992), anti-p-AKT (1:1000, Affinity, AF0016), anti-AKT (1:1000, Affinity, AF0836), anti-VEGFR2 (1:1000, Affinity, AF6281), anti-β-actin (1:2000, Proteintech, Wuhan, China, #60008-1-Ig), goat anti-mouse IgG-HRP (1:10000, Proteintech, #SA00001-1), goat anti-rabbit IgG-HRP (1:10000, Proteintech, #SA00001-2).
RNA isolation and real-time quantitative PCR (RT-qPCR) analysis
Total RNA was isolated using TRIpure reagent (BioTeke, Beijing, China) in strict accordance with the manufacturer’s instructions. All extracted RNA was subjected to Nanodrop 2000 (ThermoFisher) to verify sample concentration and purity before cDNA synthesis. The cDNA was synthesized using M-MLV reverse transcriptase (Beyotime) in a total 20 μL reaction buffer containing random primers, oligo (dT)15 primers at 25 °C for 10 min, 42 °C for 50 min, and 80 °C for 10 min. The prepared cDNA samples were tested to examine the expression of genes using Exicycler™ 96 Real-Time Quantitative system (BIONEER, Daejeon, Korea) in the presence of SYBR Green Master Mix (Solarbio). Primers sequences from 5′ to 3′ (synthesized by GenScript, Nanjing, China) were listed as follow: homo-AURKA (ACCTTCGGCATCCTAATA and AGCATGTACTGACCACCC); homo-VEGFA (TCACCAAGGCCAGCACATAG and GGGCACCAACGTACACGCT); homo-GPX4 (GAAGCAGGAGCCAGGGAGT and ACGCAGCCGTTCTTGTCG); homo-SLC7A11 (CCCTTTCCCTCTATTCGG and ACCTGGGTTTCTTGTCCC); homo-ACSL4 (GCATTCCTCCAAGTAGACC and ATGAGCCAAAGGCAAGT); homo-ALOX5 (GACAAGCCCTTCTACAACGA and CCATCCCTCAGGACAACC); mus-AURKA (CTTTCCCTGACTTTGTGAC and ATTTGCTGGTTGGCTCTT); mus-GPX4 (CAACCAGTTTGGGAGGC and CTTGGGCTGGACTTTCAT); mus-SLC7A11 (TTGGAGCCCTGTCCTATGC and CGAGCAGTTCCACCCAGAC).
Detection assay kit
The content of malondialdehyde (MDA) (in cells or gastrocnemius muscles), glutathione (GSH) (in cells or gastrocnemius muscles), hemoglobin (in matrigel plug), and VEGFA (in gastrocnemius muscles) was detected using corresponding commercial detection kit or ELISA kit, following the manufacturer’s recommendation. Cell cycle detection was conducted using commercial cell cycle detection kit (KeyGEN) according to protocol’s instruction by flow cytometer.
Statistics analysis
Statistical analyses were conducted using Graphpad Prism 8.0. All results are shown as scatter and mean ± standard deviation (SD), except behavioral deficits scores (each dot represented the corresponding scores per animal). Each experiment contained at least three biological replicates, of each performed in triplicate. One- and two-way analysis of variance (ANOVA) with Tukey’s post-hoc tests were utilized to analyze statistical differences between multiple groups. Student’s t-test was utilized to analyze statistical differences between two groups. P value < 0.05 was recognized as significance (*P < 0.05, **P < 0.01).



留言 (0)