
記住我


In this study, CLDN6 was chosen to be evaluated as therapeutic targets by using ADC to treat GCT cells.
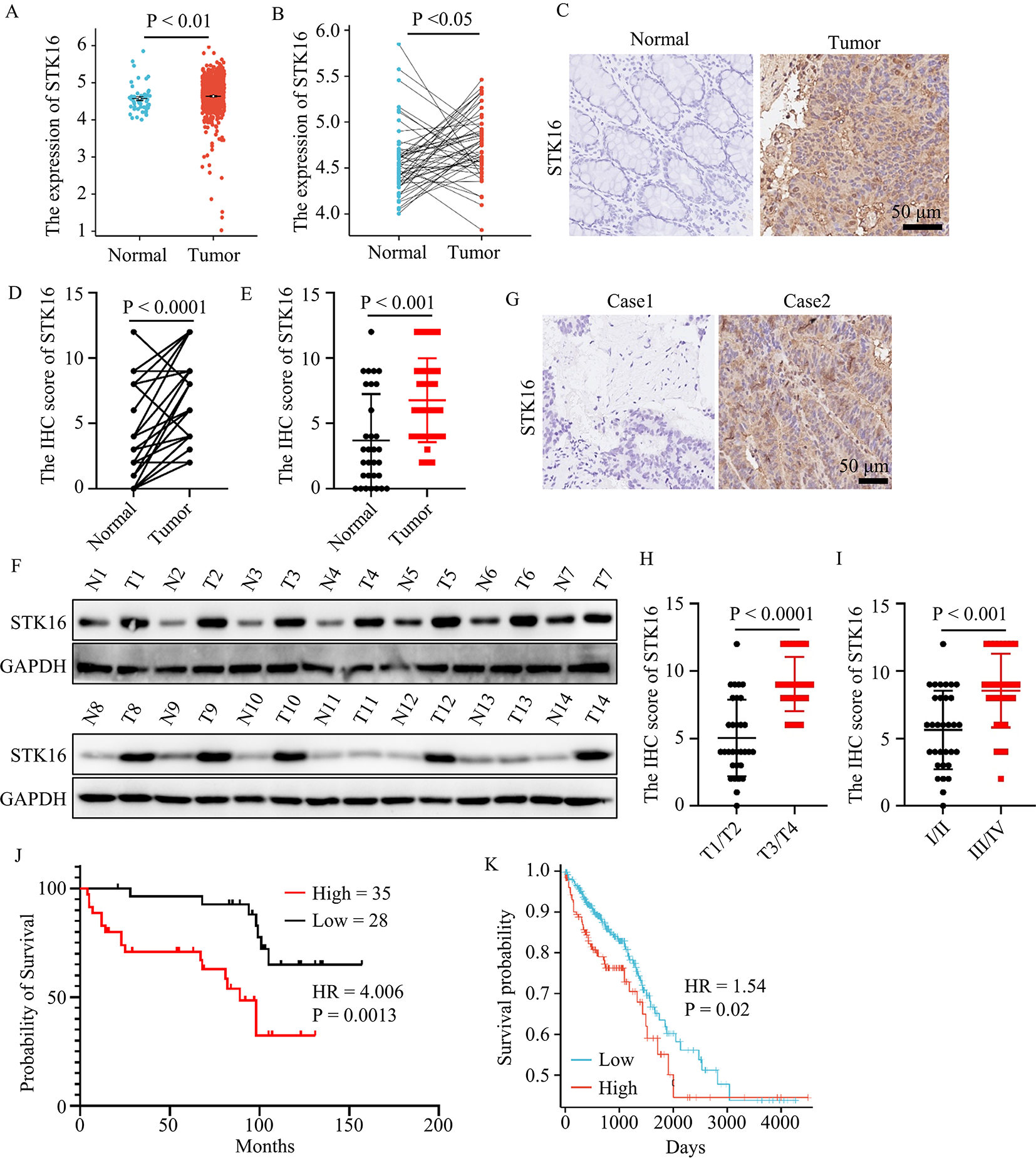
While genomic alterations in CLDN6 were merely observed in the TCGA GCT cohort, we could further show CLDN6 / CLDN6 being detectable on mRNA and protein level in GCT cell lines including cisplatin-resistant subclones (-R) derived from SEM (TCam-2), EC (2102EP, NCCIT, NT2/D1), CC (JAR, JEG-3, BeWo), and an EC-YST-intermediate (1411H) (Additional file 1: Fig. S1A; Fig. 1A, B). In male YST cells (GCT72(-R)), a predominant CLDN6− and a small CLDN6+ population was found, while only low levels of CLDN6 / CLDN6 were observed in the female YST cell line NOY-1(-R), which were comparable to those of non-cancerous control cells (i. e. fibroblasts, immune cells, keratinocytes) (Fig. 1A, B). Next, the effects on cell viability, apoptosis rates, and the cell cycle distribution of the novel CLDN6-ADC were evaluated by XTT assays and flow cytometry, respectively (Fig. 1C, D). Treatment with the CLDN6-ADC reduced cell viability (LD50 72 h 191 - 641 ng / ml) and induced apoptosis in most CLDN6+ GCT(-R) cells (i. e. SEM, EC, CC, and YST cell lines) in comparison to the monoclonal antibody alone (Fig. 1D; Additional file 1: Fig. S1B). After 48 h, EC(-R) cell lines showed the strongest increase in apoptosis rates, while male YST cell lines (GCT(-R)) showed only a mild increase in apoptosis and no alterations in the cell cycle phase distribution (Fig. 1C, D). Female CLDN6− NOY-1 as well as non-cancerous control cells did not respond to CLDN6-ADC treatment (Fig. 1D), while MMAE alone expectedly reduced cell viability in GCT cells at low concentrations (LD50 72 h 0.19 - 10.7 nM) (Additional file 1: Fig. S1B). Moreover, the CLDN6-ADC caused mainly accumulation in the G2 / M cell cycle phase in CLDN6+ cells (TCam-2(-R), 2102EP, NCCIT(-R), JAR, JEG-3(-R), 1411H), but not in CLDN6− cells (Fig. 1C, Additional file 1: Fig. S1C). Similar to the treatment with MMAE alone, also mitotic catastrophes were observed upon treatment with the CLDN6-ADC (NT2/D1(-R)) (Fig. 1C, Additional file 1: Fig. S1C). Therefore, the CLDN6-ADC is suitable for the treatment of the GCT subtypes SEM, EC, and CC. In male YST cells, the CLDN6-ADC is less efficient compared to the other GCT entities, while the ADC is not suitable to target female YST cells. In fact, this observation was further validated via immunohistochemical stainings of CLDN6 in YST-R tissues (n = 10), where only 40 % of the investigated cases presented as CLDN6+ (Additional file 1: Fig. S1D).
Fig. 1
CLDN6-ADC as a novel therapeutic option to target GCTs. A Raw flow cytometry data of CLDN6-FITC stained (blue) GCT cell lines, including their cisplatin-resistant sublines, and non-cancerous control cells compared with unstained controls (grey). B Relative CLDN6 expression in GCT cell lines and non-cancerous control cells. ACTB and GAPDH were used as housekeeping genes. C LD50 values (ng / ml) acquired by XTT cell viability assays 72 h after treatment with CLDN6-ADC and color-coded changes in cell cycle distribution (G2 / M = green, mitotic catastrophe = red, changes < 5 % = grey) upon treatment with CLDN6-ADC as compared to treatment with the CLDN6 antibody alone in GCT cell lines, including their cisplatin-resistant sublines, as well as fibroblast control cells (MPAF). D Lollipop graph summarizing relative number of apoptotic cells in GCT cell lines and fibroblast control cells after treatment with either CLDN6-ADC or CLDN6 antibody alone
Identification of novel therapeutic options for YSTSince CLDN6 levels were rather low in YST cells, representing the most aggressive and persistent GCT subtype, eventually, the CLDN6-ADC showed only a moderate efficiency in YST cells. Hence, we characterized therapy-resistant YST to identify putative therapeutic targets, which can be attacked by multikinase inhibitors.
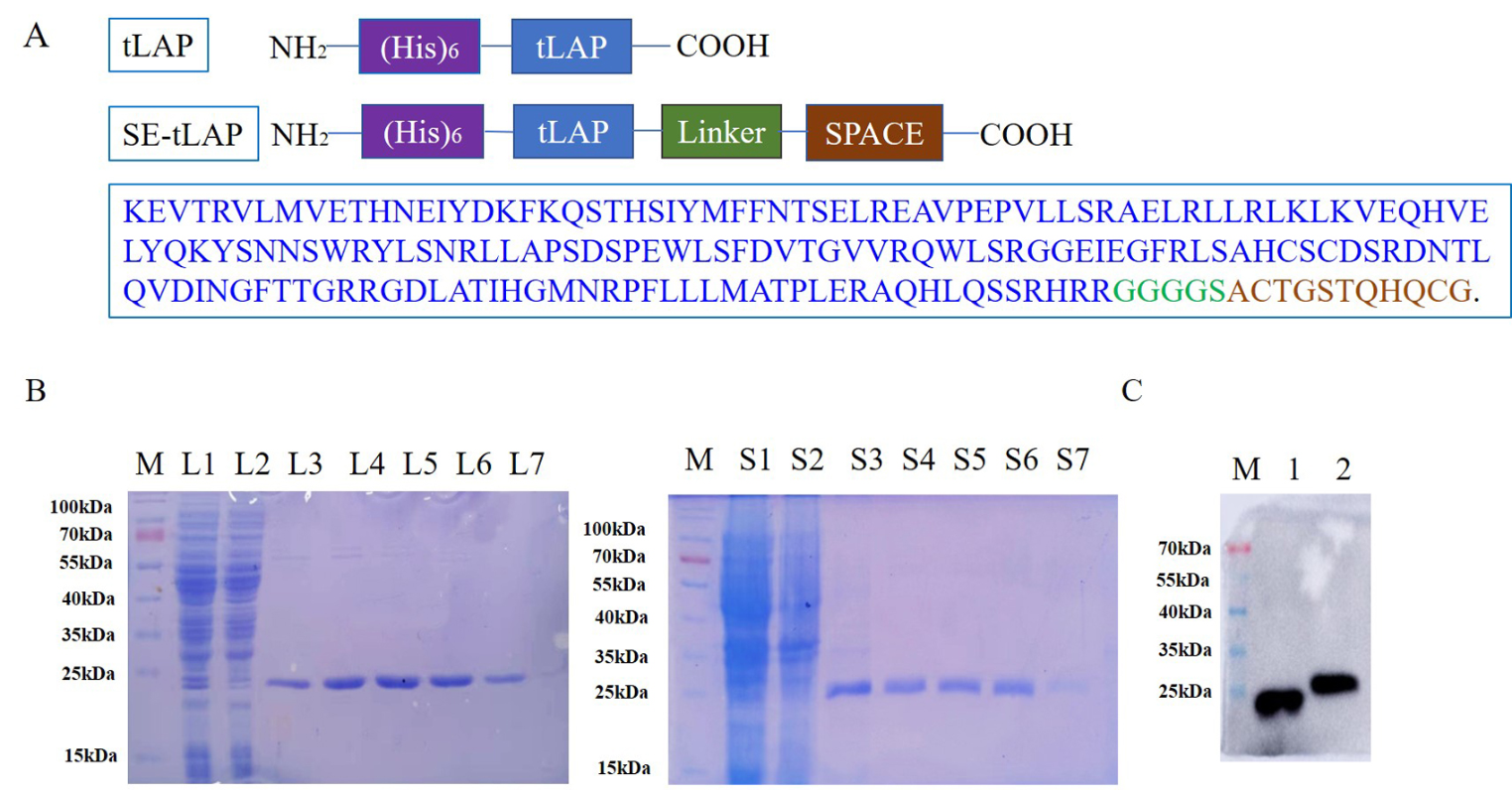
TSO analyses of refractory YST (YST-R) tissues (n = 6) were performed to identify druggable genomic alterations. We detected a mean of 3.2 mut / Mb (0.8 - 5.6 mut / Mb) in the YST-R samples, though, the tumor mutational burden (TMB) did not correlate to the microsatellite instability score (MSI; 3.7 % (1.67 - 6.36 %)) (Fig. 2A, B). Single nucleotide variants (SNV) in TP53 (c.215C > G), BRCA2 (c.7397T > C), IL7R (c.197T > C, c.412G > A), and SPTA1 (c.5077A > C) were observed in all YST-R samples. Furthermore, CHEK1, FGF6, FGF23, and KRAS were amplified, but with a low fold change (max 2.2), and SNVs were detected in FGFR4 (c.1162G > A), KMT2A (c.10841T > C), NTRK1 (c.53G > A, c.1810C > T, c.1838G > T), and TSC2 (1747G > A, c.4285G > T) in at least 50 % of the evaluated samples (Fig. 2C; Additional file 6: Data S1A). Additionally, besides further SNV, amplifications of ALK, ATM, CDK4, CHEK2, FGFR1, MDM4, and MYCN were observed in individual samples (Fig. 2C; Additional file 6: Data S1B). Hence, tumor suppressors and DNA repair key players, as well as factors related to the cell cycle, actin skeleton, or the MAPK and FGF signaling pathways were frequently altered in YST-R. It has to be noted that most found mutations were SNV classified as ‘conflicting_interpretations_of_pathogenicity’, suggesting that further work is necessary to narrow down the consequences of these mutations, with the exception of TP53 and FGFR4, whose SNV were classified as affecting ‘drug response’ and ‘pathogenic’, respectively (Additional file 6: Data S1A).
Fig. 2
Mutational profiling of YST-R. A Tumor mutational burden score (TMB) and microsatellite instability score (MSI) found in six cisplatin-resistant YST samples. B Pearson’s correlation plot of TMB and MSI. C List of identified individual and common genomic alterations found in six cisplatin-resistant YST tissues
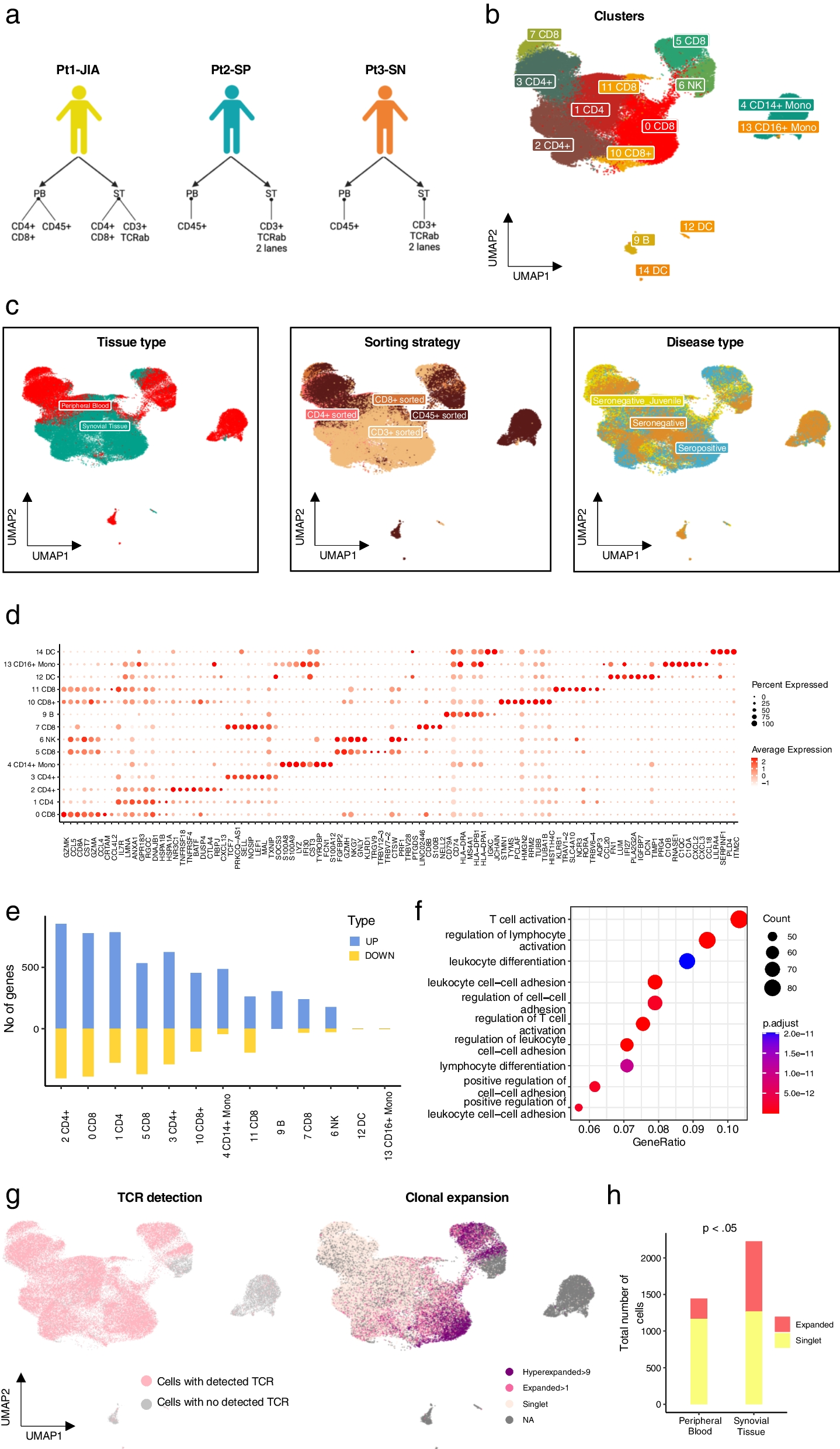
A phospho-kinase array of YST-like cells (GCT72, NOY-1(-R), 1411H) has been performed to identify signaling molecules and putative YST-specific targets, which were not present in non-cancerous control cells (MPAF) (Additional file 3: Fig. S3A). Compared to fibroblasts, in YST cell lines high levels of AKT1 - 3 (T308, S473), ERK1 / 2 (T202 / Y204, T185 / Y187), GSK3α / β (S21 / S9) and p53 (S15, S46, S392) phosphorylation were detected (Fig. 3D, Additional file 2: Fig. S2 A).
Fig. 3
Identification of novel targets for the treatment of YST. A Densitometric evaluation of absolute pixel intensities of the 13 most prominent phosphorylation sites in cell lysates from GCT72, 1411H, NOY-1, and MPAF, as measured by the human phospho-kinase array. B Graphical illustration of potential pharmacological inhibitors based on genomic alterations found in at least 50 % of YST-R samples and changes on protein level. C LD50 values (72 h) of GCT72(-R), 1411H, NOY-1(-R), and MPAF upon treatment with the inhibitors selected in (B). Inhibitors showing LD50 values below 5 µM (green) in GCT72 and higher LD50 values in MPAF (5 - 10 µM = yellow, > 10 µM = red) were further evaluated. D Color-coded changes in cell cycle distribution (G1 = light blue, S = yellow, G2 / M = green, mitotic catastrophe = red, changes < 5 % = grey) upon treatment with LD50 (72 h) concentrations for 24 h with indicated drugs, as compared to the solvent control (DMSO) in GCT72(-R), 1411H, NOY-1(-R), and MPAF. E Lollipop graph summarizing relative number of apoptotic cells in GCT cell lines and fibroblast control cells after treatment with LD50 (72 h) concentrations for 48 h with the indicated drugs in comparison to the solvent control. Of note, due to high autofluorescence of Nintedanib, all cell types were treated with LD50 values (72 h) of GCT72. F Densitometric evaluation of relative pixel intensities of the most prominent phosphorylation sites in cell lysates from GCT72 and GCT72-R treated with AZD7762, Danusertib, Nintedanib, OSU-03012, or SNS-314 (24 h, LD50 72 h) in comparison to the solvent control (DMSO), as evaluated by the human phospho-kinase array
Thus, based on the TSO and phospho-kinase array, we included AZD4547 (FGFR1-4), Nintedanib (FGFR1 - 3), AZD7762, MK-8776 (both CHEK1), and Rapamycin (mTOR) as potential inhibitory drugs to target YST(-R) (Fig. 3B). Additionally, based on the previously described resemblances between YST and HCC, we included drugs to treat HCC, i. e. Sorafenib, Lenvatinib, Regorafenib, and Cabozantinib (Fig. 3B) (Fonseca et al. 2020). The mRNA expression levels of the putative targets of these (multikinase) inhibitors were evaluated in GCT72, 1411H, NOY-1, and MPAF cells. Here, AURKA / B, CSK, FGFR1 / 2, KIT, PARP1 / 2, PDGFRA, RAF1, and YES1 were specifically expressed in the YST cells in comparison to fibroblasts (Additional file 2: Fig. S2B). The mutational status as well as mRNA expression level of respective targets of the putative (multikinase) inhibitors were also evaluated in the TCGA GCT cohort (Additional file 3: Fig. S3). No aberrations were observed in ABL2, AURKA, AURKB, AXL, BRAF, BTK, FGFR2 / 3, MET, PDGFRB, REG1A, and SRC, and only few deep deletions (0.7 %) were noted in ABL1, FGFR1 / 4, FLT4, MTOR, PARP, PDK1, RET, while CSK, EGFR, ERBB2, and RAF1 harbored missense mutations (Additional file 3: Fig. S3 A). PDGFRA amplifications and CHEK1 deletions were noted in 2.1 % and 8.0 % of GCT cases, respectively. Missense mutations as well as amplifications in KIT were observed in 15 % of the GCT patients, though mostly in SEM (Additional file 3: Fig. S3A). Regarding the mRNA levels of these putative targets, specific expression profiles / clusters were noted. As such, most SEM tissues showed specifically high levels of BTK, CSK, FGFR3, KIT, ABL2, RET, PARP2, RAF1, and PDK1, while non-seminomatous GCT were positive for AURKA / B, FGFR1 / 4, YES1, MET, ERBB2, EGFR, FLT4, PDGFRA / B, AXL, and SRC (Additional file 3: Fig. S3B). High expression levels of ABL1, FGFR2, MTOR, BRAF, and PARP1 were seen in both tumor subtypes (Additional file 3: Fig. S3B).
Out of the 17 tested multikinase inhibitors, treatment of GCT72 cells with Danusertib, SNS-314 (both AURKA - C), Nintedanib (VEGFR1 - 3, FGFR1 - 3, PDGFRA / B), Sorafenib (RAF1, BRAF, VEGFR2 / 3, PDGFRB, FLT3, KIT), Talazoparib (PARP1 / 2), OSU-03012 (PDK1), AZD4547 (FGFR1 - 3), AZD7762 (CHEK1), or Rapamycin (mTOR) resulted in LD50 values of below 5 µM (Fig. 3C, Additional file 4: Fig. S4A, B). All other drugs showing higher LD50 values were excluded from further analyses (Fig. 3C, Additional file 4: Fig. S4 A, B). Effects on cell viability upon treatment with the most potent inhibitors were further evaluated in NOY-1(-R) (YST), 1411H (EC-YST-intermediate), and fibroblasts (MPAF). With the exception of Nintedanib, LD50 values in fibroblasts upon treatment with these drugs were above 5 µM, thereby offering a therapeutic window (Fig. 3C, Additional file 4: Fig. S4B). Next, the cell cycle distribution as well as apoptosis induction upon treatment with AZD4547, AZD7762, Danusertib, Nintedanib, OSU-03012, Rapamycin, SNS-314, Sorafenib, and Talazoparib were evaluated in the four YST-like cell lines (GCT72, NOY-1(-R), 1411H) and fibroblast controls (MPAF) (Fig. 3D, Additional file 4: Fig. S4C). In comparison to the solvent control (DMSO), treatment with AZD7762, Danusertib, OSU-03012, SNS-314, Sorafenib, and Talazoparib affected the cell cycle in most GCT cells in a cell line-dependent manner. Prominently, treatment with Danusertib, SNS-314, or Talazoparib resulted in a mitotic catastrophe in YST-like cells after 24 h, while fibroblasts were only affected slightly, showing a small accumulation in the G0 / G1 or G2 / M phase upon treatment with AZD4547 or SNS-314, respectively (Fig. 3D, Additional file 4: Fig. S4C).
Of the four tested YST-like cell lines, the GCT72 and 1411H showed the highest apoptosis induction under most conditions, while the female NOY-1(-R) were the least sensitive YST-like cells (Fig. 3E). Induction of apoptosis remained rather low (< 5 %) in fibroblasts (Fig. 3E). Taking together, treatment with AZD4547 and Nintedanib resulted in apoptosis induction without altering the cell cycle distribution, while treatment with AZD7762, Danusertib, SNS-314, Sorafenib, and Talazoparib not only disrupted the cell cycle, but also induced apoptosis specifically in GCT72 YST cells (Fig. 3D, E; Additional file 4: Fig. S4C). Subsequently, the molecular effects upon treatment with the most sensitive multikinase inhibitors AZD7762, Danusertib, Nintedanib, OSU-03012, and SNS-314 have been evaluated in GCT72(-R) cells (Fig. 3F, Additional file 2: Fig. S2C, D). As such, treatment with the CHEK1 inhibitor AZD7762 enhanced phosphorylation of CHEK2 (T68), while it decreased activity of GSK3α / β (S21 / S9), SRC (Y419), STAT5a / b (Y694 / Y699), and WNK1 (T60) in GCT72(-R) cells. Danusertib treated cells presented elevated phosphorylation of GSK3α / β (S21 / S9) and p53 (S46, S392), while HSP60 and phosphorylation of ERK1 / 2 (T202 / Y204, T185 / Y187), SRC (Y419), and WNK1 (T60) were diminished in both cell lines. Nintedanib treatment resulted commonly in both cell lines in decreased levels of HSP60 and phosphorylation of p53 (S15, S392), SRC (Y419), and WNK1 (T60). Treatment with the PDK1 inhibitor OSU-03012 resulted in several shared abundances on phospho-proteome level in both cell lines (increase in CREB (S133), ERK1 / 2 (T202 / Y204, T185 / Y187), GSK3α / β (S21 / S9) activity), however, phosphorylation of p38α (T180 / Y182), p53 (S15, S46), PRAS40 (T246), STAT3 (Y727), and WNK1 (T60) were oppositional in GCT72(-R) cells. As an AURKA / B inhibitor, treatment with SNS-314 led to increased activity of GSK3α / β (S21 / S9) and p53 (S46, S392) in both cell lines. Additionally, decrease in YES (Y426) phosphorylation was found in both cell lines upon treatment with all here tested inhibitors. Remarkably, all five inhibitors resulted in diminished phosphorylation of AKT1 - 3 (S473) and WNK1 (T60) specifically in the resistant cell line, thereby indicating that the PI3K / PDK1 signaling cascade might be putatively targetable in YST-R (Fig. 3F, Additional file 2: Fig. S2C, D).
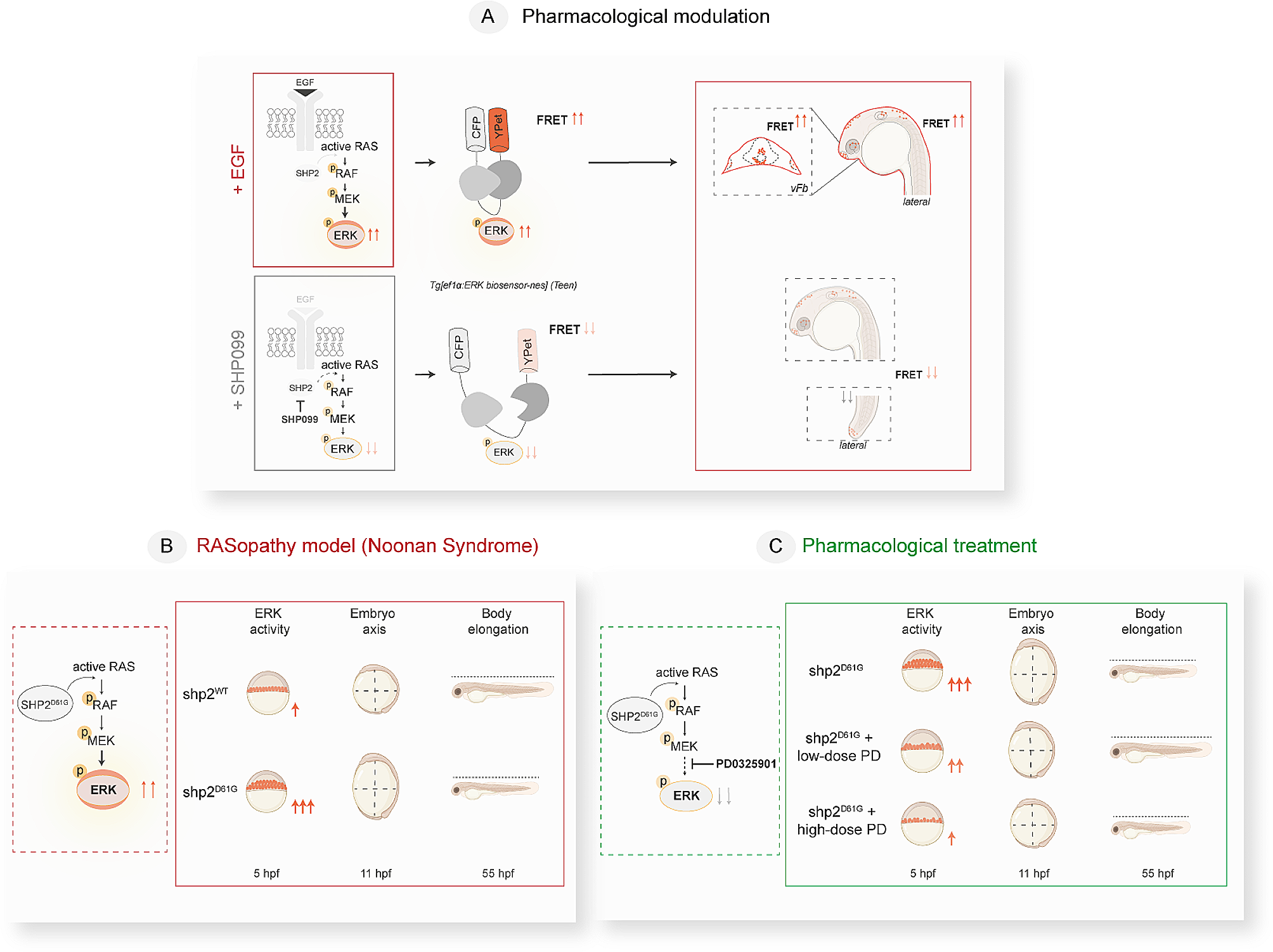
Consequently, based on a molecular-guided approach, the authors could identify suitable YST-specific candidates that could be targeted using already tested or even approved multikinase inhibitors. Nevertheless, one of the major obstacles during cisplatin-based chemotherapy of YST is the development of resistance mechanisms. At present, this fundamental process is poorly understood. Thus, we subsequently aimed at the molecular characterization of potential mechanisms driving towards a resistant phenotype in YST. Analyzing the differences between YST cells and their resistant sublines, elevated phosphorylation of AKT1 - 3 (S473) and p53 (S15, S46) were seen in GCT72-R cells, while activity of ERK1 / 2 (T202 / Y204, T185 / Y187), FGR (Y412), GSK3α / β (S21 / S9), p38α (T180 / Y182), p53 (S392), PDGFRβ (Y751), SRC (Y419), STAT5a / b (Y694 / Y699), WNK1 (T60), and YES (Y426) was reduced in the resistant subline (Fig. 4A, Additional file 2: Fig. S2C). To further decipher the underlying molecular mechanisms driving YST to a resistant phenotype and identify further targets for the treatment of YST, mass spectrometry-based proteome analyses were performed. A PCA revealed that YST-R samples (n = 5) clustered distinguishably apart from primary YST tissues (n = 9) (Fig. 4B). Even though both tissue types had a high correlation (r2 = 0.95) (Fig. 4C), 84 proteins were highly enriched and 67 were significantly depleted (abundance ratio < 0.5 or > 2, p-value < 0.05) in YST-R compared to therapy-naïve YST (Fig. 4D, Additional file 6: Data S1C). A DAVID-based gene ontology and STRING interaction analysis of the proteins enriched in YST-R (abundance ratio > 2) revealed that these factors are involved in translational initiation and RNA binding (e. g. RPL34, RPS8, EIF3G), extracellular matrix (ECM)-related processes (e. g. COL3A1, COL2A1, ITGAX, MFAP5), as well as innate / humoral (pathogen-dependent) immune response (e. g. CD36, HLA-DPB1, HLA-DRB1, LILRB5). Additionally, factors relevant during oxidative stress response and MAPK / Ras / Rap1 signaling (RALB, RAP1A, GNG12, DUSP9, PPP5C) were identified as putative supporting processes in the acquisition of resistance in YST (Fig. 4E, F).
Fig. 4
The molecular resistance mechanisms in YST. A Densitometric evaluation of relative pixel intensities of the most prominent phosphorylation sites in cell lysates from GCT72-R cells in comparison to the parental cell line, as evaluated by the human phospho-kinase array. B PCA plot, C Pearson’s correlation plot and D Volcano plot of mass-spectrometry data of FFPE-embedded YST(-R) tissues. E Enrichment plots showing gene ontology terms found exclusively in YST-R tissues. F STRING interaction analysis of YST-R-specific proteins
留言 (0)