
記住我

Idiopathic inflammatory myopathies (IIMs) are a group of relatively rare systemic autoimmune disorders with an estimated global prevalence of 2.9 to 34 per 100,000 individuals [1]. IIMs have traditionally been classified into five major subgroups based on their clinicopathologic characteristics: dermatomyositis (DM), polymyositis (PM), immune-mediated necrotizing myopathy (IMNM), anti-synthetase syndrome (ASyS), and inclusion-body myositis [2]. Most of the IIMs are characterized by symmetric proximal muscle weakness and chronic inflammation [1, 2]. DM is typically accompanied by characteristic skin manifestations, such as heliotrope rash and Gottron’s sign or papules [3]. Extramuscular manifestations include dysphagia, arthritis [4], interstitial lung disease (ILD) [5], cardiovascular effects (e.g., myocarditis) [6], and Raynaud’s syndrome [7]. In addition, myositis-specific autoantibodies are present in approximately 50–70% of patients with DM and PM [8, 9].
Classification criteria have been accepted by the American College of Rheumatology (ACR)/European League Against Rheumatism (EULAR) that allow clinicians to distinguish IIM from conditions with similar phenotypes and to subclassify patients into the major IIM subgroups [10]. However, there remains a lack of standardized therapeutic guidelines [11, 12], and some patients cannot tolerate or do not adequately respond to standard-of-care therapies. Further, some specific manifestations refractory to standard immunosuppressive therapies are a significant challenge for clinicians, including calcinosis universalis, aphagia, rapidly progressive interstitial lung disease, and chronic intestinal pseudo-obstruction [12]. This complicates the disease landscape and highlights the need for alternative therapies for these patients with refractory disease.
Here we present the clinical evidence for the safety and efficacy of Acthar® Gel (repository corticotropin injection) and how it may be considered as an alternative treatment of DM and PM. This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Epidemiology, Diagnosis, and Pathophysiology of DM and PMDM and PM are associated with increased risk of morbidity and mortality [13]. The most common causes of death are cancer, infection, profound effects of muscle weakness, and cardiovascular disease [14]. Both DM and PM have been shown to be more prevalent in female and Black/African-American patients [15,16,17,18].
The diagnoses of DM and PM are based on clinical signs and symptoms, muscle biopsies to identify inflammatory features, electromyography for evaluating myopathic vs. neuropathic causes of weakness, magnetic resonance imaging to identify active inflammation in muscle or fascia, and serum levels of muscle-derived enzymes (e.g., creatine kinase) [2]. Myositis-specific autoantibodies are generally mutually exclusive and can be associated with specific disease manifestations that help guide classification, prognosis, and disease management [8, 9].
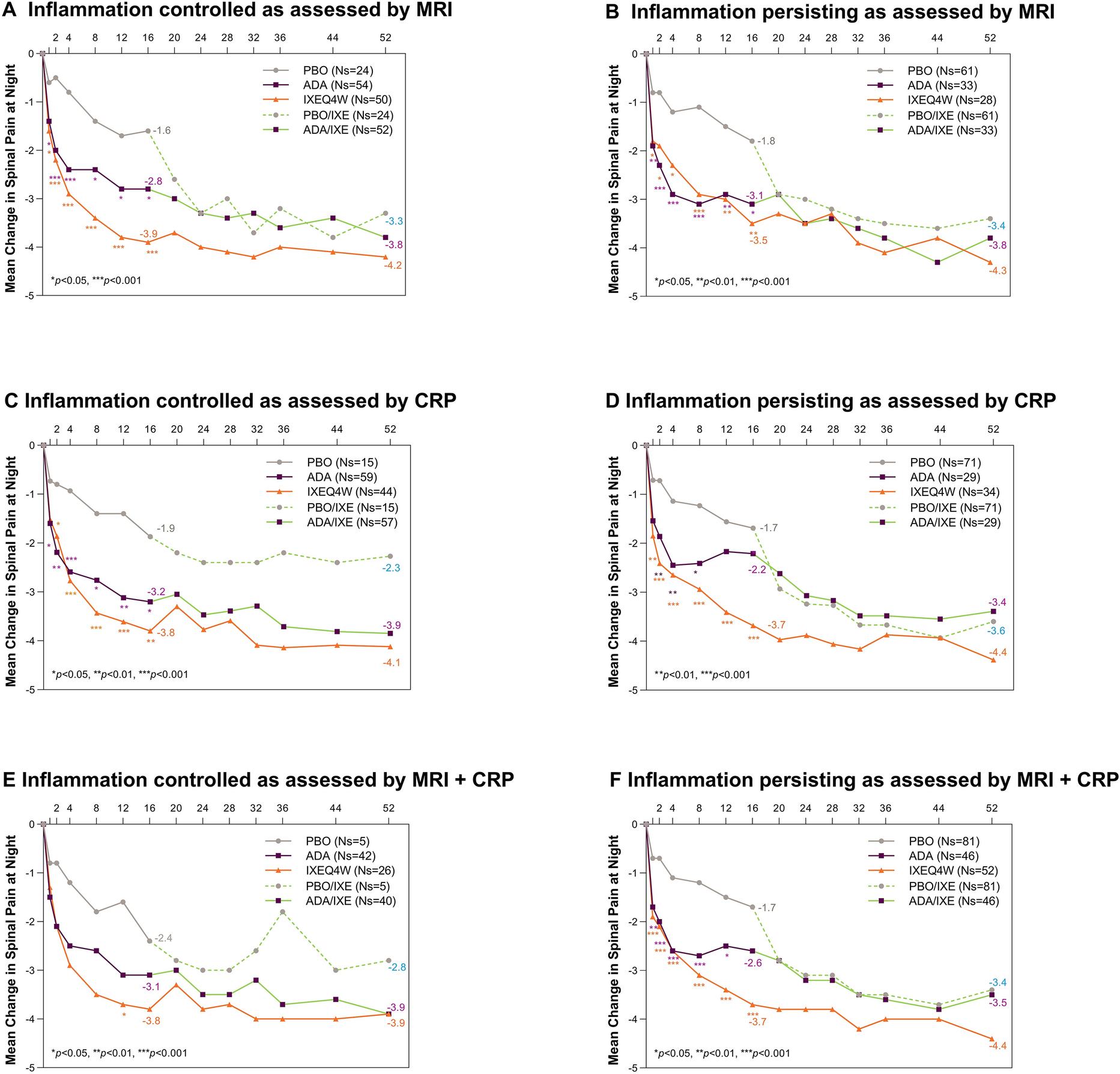
DM is characterized by perimysial and perivascular inflammation and muscle fiber atrophy (Fig. 1) [2]. It begins when putative antibodies directed against endothelial cells of the endomysial capillaries activate the classical complement cascade. This results in the formation and deposition of the membrane attack complex (MAC) on capillaries surrounding the muscle fibers and leads to endothelial cell death and ischemic muscle fiber damage [19,20,21]. Complement activation also leads to the release of proinflammatory cytokines and chemokines that upregulate the expression of adhesion molecules on the endothelial cell membrane, facilitating the transmigration of activated CD4+ T cells, B cells, and macrophages to the endomysial tissue [19,20,21]. Perivascular, perimysial, and perifascicular inflammation, along with perifascicular atrophy and reduced capillaries, are characteristic of the muscle biopsy in DM [2]. This, along with the skin manifestations of heliotrope rash, Gottron’s papules, or Gottron’s sign, provides the basis for DM diagnosis [22, 23].
Fig. 1
Pathophysiology of dermatomyositis. Muscle fiber atrophy begins when putative antibodies directed against endothelial cells of the endomysial capillaries activate the classical complement cascade. The membrane attack complex (MAC) is deposited on capillaries surrounding the muscle fibers and leads to endothelial cell death and ischemic muscle fiber damage. Complement activation also leads to the release of proinflammatory cytokines and chemokines that upregulate the expression of adhesion molecules (VCAM-1, ICAM-1) on the endothelial cell membrane, facilitating the transmigration of activated CD4+ T cells, B cells, and macrophages to the endomysial tissue. Figure created with BioRender.com. C1 indicates complement component 1; C3, complement component 3; C3b, complement component 3b; ICAM-1, intercellular adhesion molecule 1; LFA-1, lymphocyte function-associated antigen 1; MAC, membrane attack complex; Mac-1, macrophage-1 antigen; pDC, plasmacytoid dendritic cell; VCAM-1, vascular cell adhesion protein 1; VLA-4, very late antigen-4
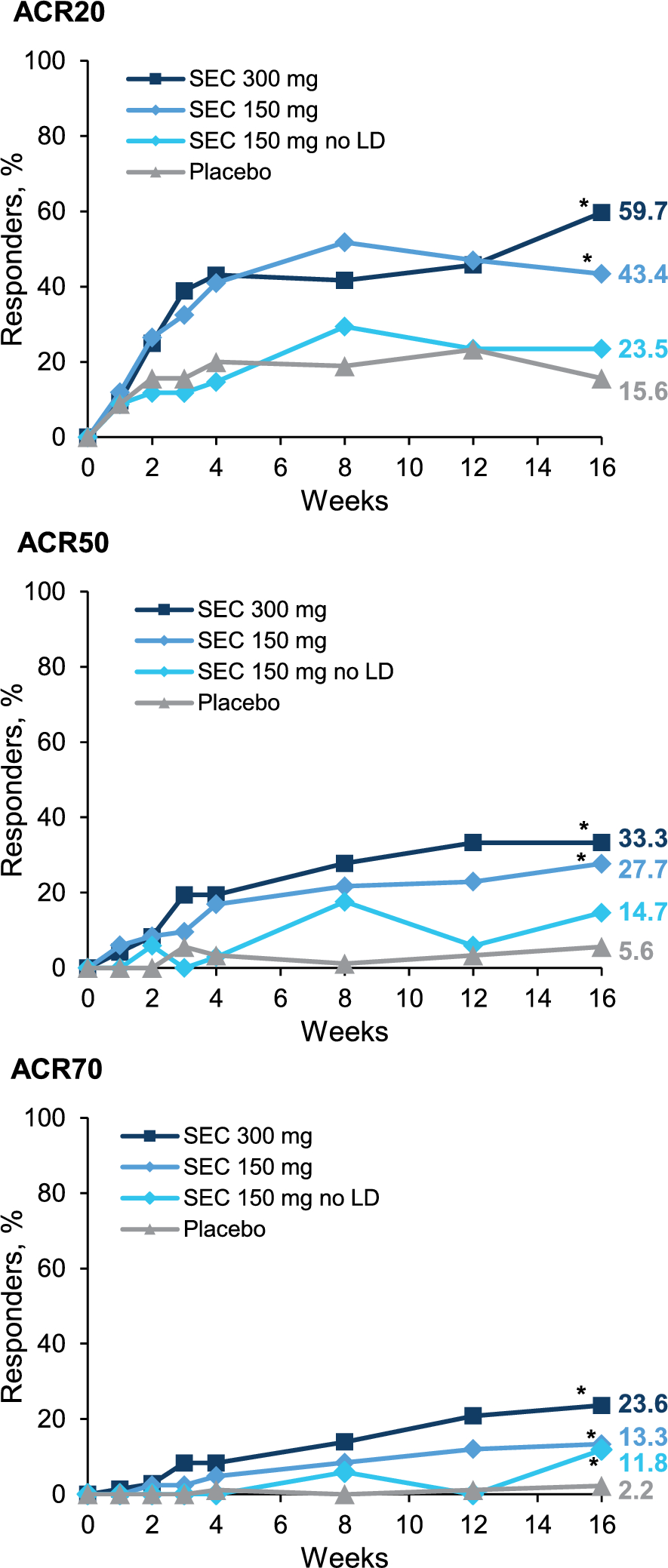
PM is characterized by endomysial inflammation [2] and muscle fiber degeneration [1]; however, the mechanism by which this occurs is not well established. Muscle biopsies from individuals with PM show an overexpression of major histocompatibility complex (MHC) class 1 antigens on muscle fibers, infiltration of CD8+ T cells, and muscle fiber necrosis (Fig. 2) [2, 19]. Muscle fiber death occurs when an antigen-specific CD8+ T cell binds to its corresponding MHC-1 expressing antigen on a muscle fiber, activating the CD8+ T cell to release perforin and granzyme granules that ultimately result in cell death [2]. Pro-inflammatory cytokines (e.g., interferon-γ, tumor necrosis factor-α) released by the activated T cells may also enhance MHC-1 upregulation and the cytotoxic effect of T cells [2]. B cells and terminally differentiated plasma cells are also found in the muscle tissue of patients with PM; however, their role in muscle inflammation has not been fully elucidated [24].
Fig. 2
Pathophysiology of polymyositis. Polymyositis is characterized by an overexpression of major histocompatibility complex (MHC) class 1 antigens on muscle fibers and the infiltration of CD8+ T cells from the periphery into the endomysium. Muscle fiber death occurs when an antigen-specific CD8+ T cell binds its corresponding MHC-1 expressing antigen on a muscle fiber, activating the CD8+ T cell to release perforin and granzyme granules and resulting in necrosis. Pro-inflammatory cytokines (e.g., interferon-γ, tumor necrosis factor-α) released by the activated T cells may also enhance MHC-1 upregulation and the cytotoxic effect of T cells. Figure created with BioRender.com. CD40 indicates cluster of differentiation 40; CD40L, cluster of differentiation 40 ligand; CD80, cluster of differentiation 80; CTLA-4, cytotoxic T lymphocyte associated protein 4; ICAM-1, intercellular adhesion molecule 1; ICOS, inducible costimulator; ICOS-L, inducible costimulator-ligand; LFA-1, lymphocyte function-associated antigen 1; MHC, major histocompatibility complex; TCR, T cell receptor; VCAM-1, vascular cell adhesion protein 1; VLA-4, very late antigen-4
The discovery of myositis-specific antibodies has allowed the identification of new subgroups of IIMs with distinct clinical and muscle histopathologic features, including IMNM and ASyS [1]. Muscle biopsies from patients with autoantibodies against aminoacyl transfer RNA synthetases, a hallmark of patients with ASyS, exhibit perifascicular necrosis [25] and endomysial infiltration by clonally expanded T cells [26]. In addition, patients with ASyS have a higher incidence of pulmonary involvement and may have glucocorticoid-resistant myositis or ILD [27]. Patients with IMNM most often have autoantibodies to 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR) or the signal recognition particle (SRP), which are highly correlated with muscle weakness and elevated creatine kinase levels [1]. Although muscle biopsies show increased macrophages and deposits of MAC on the sarcolemma, IMNM-specific autoantibodies do not appear to be complement-fixing [28], and the pathophysiological mechanism of muscle damage remains uncertain [1].
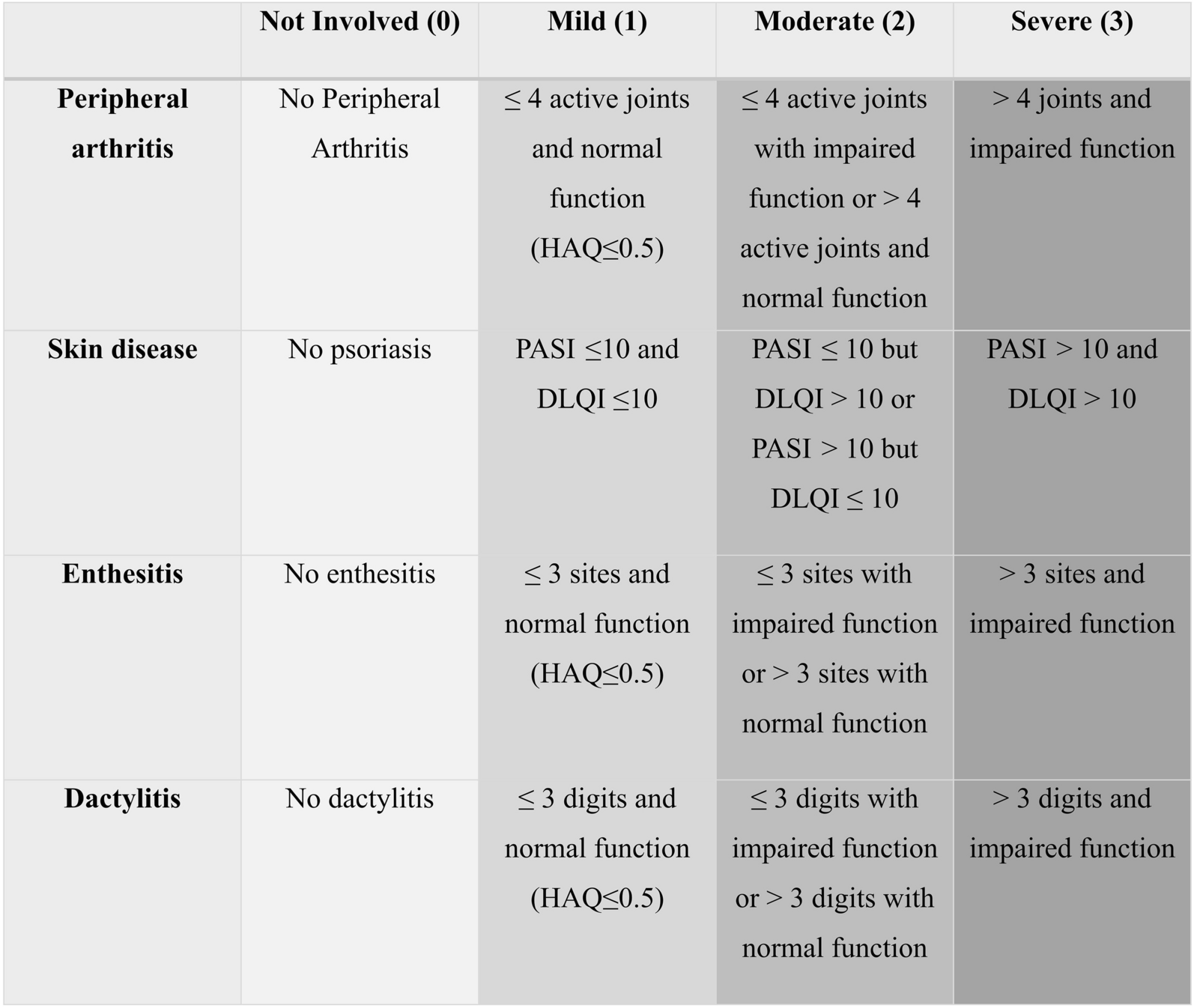
Current Standard of CareCurrent clinical practice guidelines indicate the use of physical therapy and sun protection (in patients with DM) in addition to traditional immunosuppressive pharmacotherapies [12]. It is generally agreed that glucocorticoids should be the first-line therapy for DM and PM [11, 12, 29, 30], with the addition of methotrexate or azathioprine for moderate-to-severe disease or to control disease flares when tapering glucocorticoids (Fig. 3) [1, 11, 29, 30]. Unfortunately, glucocorticoids are not an optimal long-term treatment option due to a high rate of adverse events (AEs) and other complications [11, 29] (e.g., diabetes, hypertension, dyslipidemia, osteoporosis, weight gain, gastric intolerance, mood changes, infections, cataracts, and glaucoma) [31]. Despite these side effects, drugs such as methotrexate, azathioprine, mycophenolate mofetil (MMF), and intravenous immunoglobulin (IVIg) are recommended for their steroid-sparing abilities. Typically, 30–40% of patients have disease that is refractory to glucocorticoids, with fewer than 50% of patients achieving a complete response and many experiencing significant side effects. Further, glucocorticoid tapering is often associated with disease flare, and most patients will require an additional immunosuppressive agent [13, 14].
Fig. 3
Current standard of care therapies for the management of idiopathic inflammatory myopathies. Reproduced from Oddis CV, Aggarwal R. Treatment in myositis. Nat Rev Rheumatol. 2018;14(5):279–289. doi: https://doi.org/10.1038/nrrheum.2018.42. IVIg indicates intravenous immunoglobulin; MMF, mycophenolate mofetil
MMF is typically used as a second-line therapy, except in patients with moderate-to-severe myositis associated with ILD and refractory DM rashes for whom it can be used as a first-line therapy [1, 11]. The calcineurin inhibitors cyclosporine and tacrolimus are also considered second-line therapies; however, they are typically reserved for refractory myositis with either muscle weakness or associated ILD due to toxicity concerns [1, 11, 30]. Third-line therapies include cyclophosphamide and biologic agents such as rituximab [1, 32,33,34]. Owing to the toxic effects and increased risk of malignancy at high cumulative doses of cyclophosphamide, its use is limited to severe refractory muscle weakness, rapidly progressive ILD, or systemic vasculitis [1, 30].
IVIg therapy is approved by the US Food and Drug Administration (FDA) for DM in adults and is considered safe and effective in combination with or after failure of glucocorticoids or other immunosuppressive drugs. However, IVIg treatment is associated with increased health care resource utilization due to administration in an outpatient setting [35]. In addition, it has a long administration time and potential side effects and is contraindicated in patients with a high risk of thromboembolism, so it is not routinely used as a first-line therapy unless there are features such as dysphagia [36], anti-HMGCR-related IMNM [37], active infection, severe disease, pregnancy, cancer, or ILD [1, 11, 12, 29, 30, 38].
Most current treatments are associated with toxicities that require careful monitoring [29]. In addition, many patients are unable to tolerate the side effects or are unresponsive to standard therapies [39], which poses a significant therapeutic challenge to clinicians.
Acthar GelActhar has been FDA-approved for use in patients with DM and PM since 1952 [40]; however, it is not routinely used in the treatment of IIMs [1]. Acthar is a naturally sourced complex mixture of adrenocorticotropic hormone (ACTH) analogs (a major component of which is ACTH1-39) and other pituitary peptides with a unique mechanism of action from standard of care therapies used to treat DM and PM [40]. It was originally thought that the anti-inflammatory effects of Acthar were mediated through glucocorticoid production (via activation of melanocortin receptor [MCR] 2 on adrenocortical cells), but recent studies have shown that glucocorticoid release from the adrenal cortex is relatively low with Acthar in both animals and humans, suggesting that it has a steroid-independent anti-inflammatory mechanism of action [41,42,43].
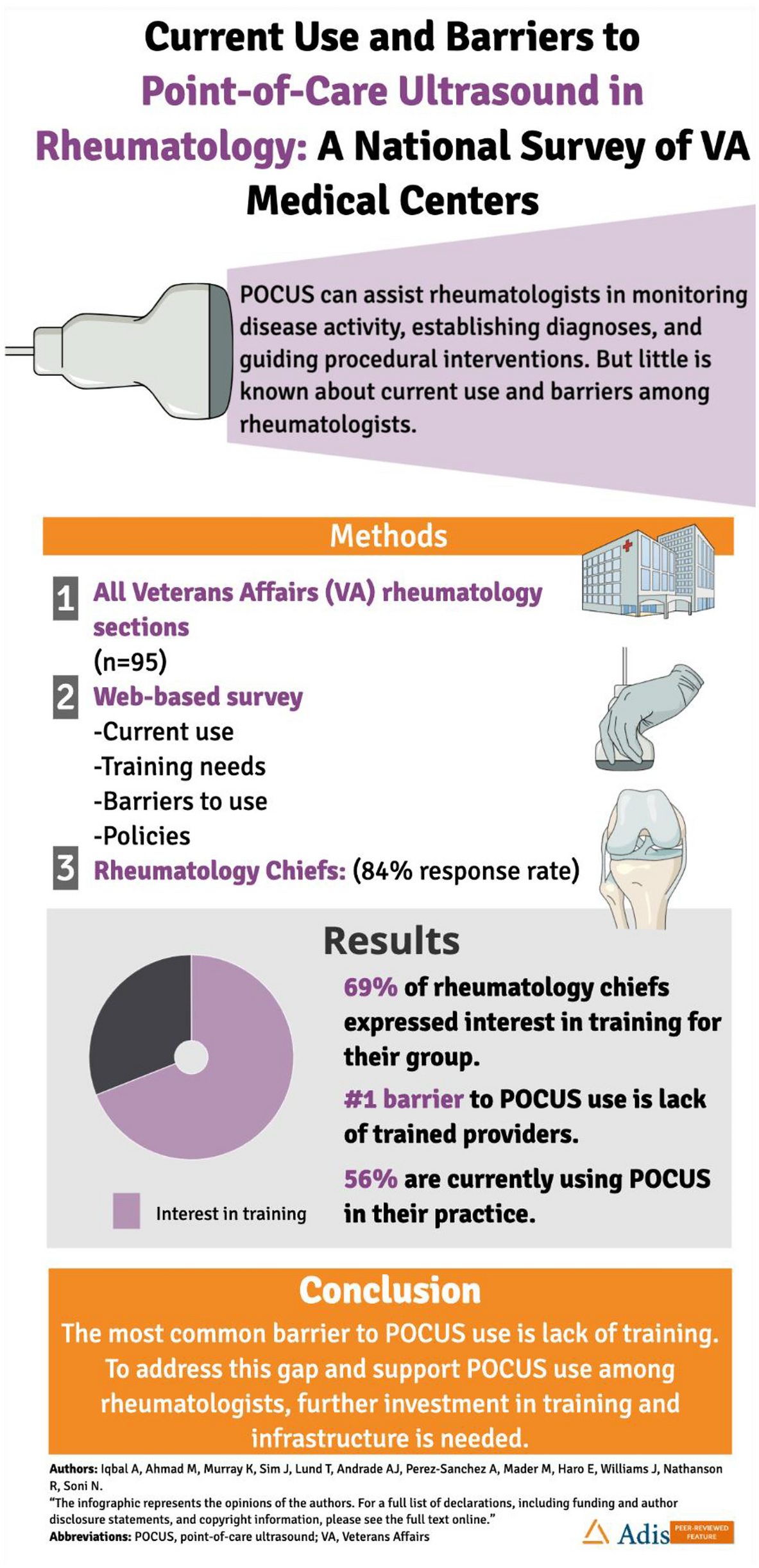
MCR agonists provide substantial anti-inflammatory and immunomodulatory effects. MCR activation inhibits nuclear factor-kappa B (NF-κB), which in turn functionally controls the expression of hundreds of genes including those that encode cytokines and their receptors, growth factors, and chemokines [44]. MC1R, MC3R, and MC5R are expressed in macrophages, B cells, and T cells and mediate anti-inflammatory and immunomodulatory properties of MCR agonists (Fig. 4) [41, 44,45,46]. Acthar has also been shown to have a direct immunomodulatory effect [41, 47, 48] via activation of MCRs, some of which are expressed on immune cells [41]. Acthar has been shown to inhibit antibody production and B cell proliferation [47] as well as inhibit inflammatory cytokine production from macrophages and T cells [48, 49].
Fig. 4
Proposed mechanism of action of Acthar. Reproduced from Mirsaeidi M, Baughman RP. Repository corticotropin injection for the treatment of pulmonary sarcoidosis: a narrative review. Pulm Ther. 2022;8(1):43–55. doi:https://doi.org/10.1007/s41030-022-00181-0. under CC BY-NC 4.0 license terms. MCR indicates melanocortin receptor
Although there have been no active-controlled studies comparing the efficacy of glucocorticoids to Acthar in DM or PM, some recent clinical trials, retrospective analyses, and case reports add to the growing body of evidence suggesting that Acthar may be effective in patients with DM and PM (Table 1). In a retrospective case review examining five patients with either DM or PM disease exacerbation who were unable to tolerate the side effects of previous therapies or in whom those therapies failed, patients received 80 U Acthar either twice weekly (n = 4) or once weekly (n = 1) for 12 weeks [50]. Improvements were observed in all patients, including increased muscle strength, decreased pain, and resolution of skin rashes in the patients with DM [50]. The success of Acthar in this small retrospective case review prompted the creation of the Acthar in Dermatomyositis and Polymyositis Treatment (ADAPT) registry in order to determine dosing, AEs, and efficacy of Acthar in patients with refractory DM or PM. An interim analysis (n = 24) showed that 58.3% of patients responded to Acthar treatment (80 IU twice weekly) as shown by improvement in inflammatory neuropathy cause and treatment (INCAT) score, manual muscle testing (MMT) scores, or Myositis Activity Profile (MAP) scores. Interestingly, the concomitant use of MMF was associated with 100% response rate (n = 5) [51].
Table 1 Summary of clinical trials, retrospective analyses, and case reports demonstrating the efficacy and safety of Acthar in patients with DM and PM patientsThe efficacy of Acthar (80 IU twice weekly) was also examined in a retrospective case series of four patients with DM or PM that was refractory to corticosteroids or other disease-modifying agents. Most patients experienced improvements in clinical laboratory measures, muscle strength, and pain. All patients were able to either decrease or stop glucocorticoid treatment following Acthar therapy [52]. In yet another retrospective analysis (n = 8) of Acthar use with doses varying between 40 IU once daily to 80 IU once a week up to 12 months in patients with DM and PM, 66.7% of patients improved based on physicians’ assessment of efficacy [
留言 (0)