
記住我
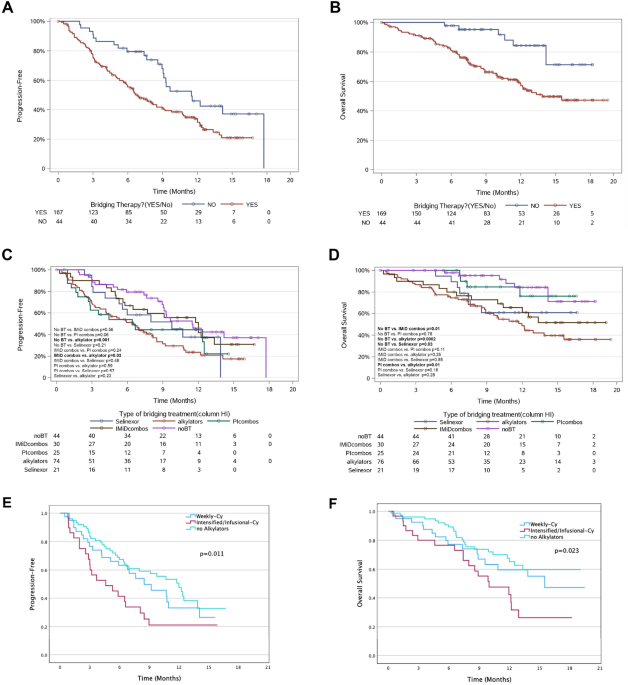
The analysis of CGAP Mitelman database [see Supplementary Table 1 in [11]] showed that ~94% of malignancies carrying del(9q34) were of hematopoietic origin, suggesting that loss of ABL1 may alter malignant transformation preferentially in the hematopoietic cells. Most of the cases carried complex karyotypes, but we identified the following chromosomal translocations in del(9q34) cases (Fig. 1A): t(8;21) AML1-ETO [t(8;21) in 14% of hematologic cases], BCR-ABL1 [t(9;22) in 10%], NUP98 translocations [t(1;11), t(2;11) and t(7;11) in 5%], CCND1 translocations [t(11;14) in 3%], BCL2 translocations [t(14;18) in 2%), MLL translocations [t(4;11) MLL-AF4 and t(10;11) MLL-AF10 in 3%], MOZ-p300 [t(8;22)] and TCF3-PBX1 [t(1;19)] (each in 1.5%). These data implicate ABL1 loss in leukemias carrying chromosomal translocations other than t(9;22) encoding for BCR-ABL1 [11]. In addition, RNA-seq analyses revealed broad spectrum of ABL1 expression in various leukemias (Fig. 1B). Overall survival of patients with leukemias in the context of ABL1 expression levels revealed that chronic lymphocytic leukemia (CLL) and AML displaying low levels of ABL1 mRNA expression had shorter overall survival, while opposite effect was detected in ALL (Fig. 1C). In addition, low ABL1 expression had a negative impact on survival of patients with small cell lung carcinoma, ovarian carcinoma, and breast carcinoma (Supplemental Fig. S1). Altogether, ABL1 expression level could be considered a prognostic factor in selected hematological malignancies and solid tumors.
Fig. 1: ABL1 as a prognostic factor.
A Chromosomal aberrations detected in leukemias carrying del(9q34) or deletions encompassing this region resulting in loss of ABL1 in CGAP Mitelman database (https://mitelmandatabase.isb-cgc.org/). B Normalized ABL1 expression from HOVON, TCGA-LAML, and MILE (left to right) datasets. HOVON and MILE datasets were sourced from Affymetrix microarrays and normalized using the RMA method, while the TCGA-LAML dataset is is a set of rlog-normalized RNA-seq expression values. C Kaplan-Meier estimates of the overall survival of patients with indicated leukemias based on ABL1 expression levels. HIGH and LOW cohorts in AML GSE22762 were divided at mean of the gene expression. Cutoffs for the AML dataset GSE37642 and the ALL datasets from TARGET ALL phases II and III (9.81, 4.05, and 10.88 of the RMA-normalized Affymetrix ABL1 expression values, respectively) were determined using the maxstat package in R, an extension of the Hothorn and Lausen (2002) method to select maximally selected cut points. p values were calculated from the log-rank test.
The top genetic aberrations in hematological malignancies, other that BCR-ABL1 were t(8;21) generating fusion protein AML1-ETO and NUP98 translocations, e.g., t(1;11) encoding NUP98-PMX1. To test the role of ABL1 in leukemogenesis mediated by these genetic aberrations Abl1−/− and Abl1 + /+ murine hematopoietic cells were employed. Since homozygous disruption of Abl1 in mice caused neonatal lethality/poor viability [19, 20], first we generated Abl1−/−;Vav-Cre (n = 19), Abl1 + /-;Vav-Cre (n = 52) and Abl1 + /+;Vav-Cre (n = 13) mice. Vav-Cre starts to express specifically in hematopoiesis system after the birth and can knock out Abl1 efficiently, which has been confirmed by PCR using peripheral blood leukocytes from the peripheral blood samples and tail tissues (Supplemental Fig. S2A, B). These mice served as bone marrow donors to study leukemic transformation.
The results clearly show that in the absence of Abl1, Lin-c-Kit+ AML1-ETO and NUP98-PMX1 cells displayed higher clonogenic activity (Fig. 2A) and gained tremendous proliferation advantage in long-term tissue culture (Fig. 2B). Conversely, the absence of Abl1 reduced the proliferation potential of normal Lin-c-Kit+ hematopoietic cells. In addition, G-CSF-induced myeloid differentiation of AML1-ETO;Abl1−/− cells was reduced when compared to AML1-ETO;Abl + /+- cells (Supplemental Fig. S2C). However, myeloid differentiation of NUP98-PMX1;Abl1−/− cells and non-transformed Abl1−/− was not significantly different from that in Abl1 + /+ counterparts (Supplemental Fig. S2D, E). This inconsistency in the impact of ABL1 on differentiation might depend on the differences between AML1-ETO and NUP98-PMX1 in regulating differentiation by downregulation of C/EBPα and c-FOS, respectively [21, 22]. This speculation is supported by the observations that C/EBPα was downregulated in BCR-ABL1 -positive cells in which loss of ABL1 attenuated differentiation [11, 23].
Fig. 2: ABL1 regulated proliferation of murine hematopoietic cells expressing AML1-ETO and NUP98-PMX1.
A, B Murine bone marrow cells from Abl1−/−, Abl + /- and Abl1 + /+ mice (3 mice/group) were infected with retroviruses and lentiviruses carrying AML1-ETO and mCherry, NUP98-PMX1 and GFP, and mCherry or GFP only. A Clonogenic activity; Insect: Western blot detecting indicated proteins using anti-AML1 and anti-NUP98 antibodies (arrow indicates the position of NUP98-PMX1 protein). Results represent mean + /- SD from triplicate experiments; p < 0.001 using Student t-test. B total number of mCherry+ and GFP + cells in liquid culture. C–E AML1-ETO and NUP98-PMX1 -positive tet-inducible ABL1-32Dcl3 cells were maintained in Tet+ or Tet- medium in the presence or absence of DPH. C Western analysis of phospho-Y412 ABL1 (p-ABL1), total ABL1, AML1-ETO, NUP98-PMX1 and actin in the indicated cells incubated or not with 10 μM DPH for 30 minutes (arrows indicate the position of AML1-ETO and NUP98-PMX1 proteins). D Clonogenic activity of AML1-ETO and NUP98-PMX1 -positive cells maintained in Tet+ medium and incubated in the presence of indicated concentrations of DPH for 4 days followed by plating in MethoCult. Results represent % of colonies ± SD when compared to DPH-untreated cells. E proliferation of AML1-ETO and NUP98-PMX1 -positive cells maintained in Tet+ medium in the presence or absence of 10 μM DPH. Results represent number of living cells detected by trypan blue exclusion ± SD.
To further explore the effect of activation of ABL1 kinase on proliferation of leukemia cells, tet-inducible ABL1-32Dcl3 cells were transduced with AML1-ETO and NUP98-PMX1 (Fig. 2C). ABL1 was overexpressed upon the treatment with tetracycline and ABL1 kinase was stimulated by DPH, an agonist of ABL1 kinase [24] (Fig. 2C). DPH-mediated activation of overexpressed ABL1 kinase was associated with reduced clonogenic activity (Fig. 2D) and proliferation (Fig. 2E) of 32Dcl3 cells.
ABL1 regulated the sensitivity of NUP98-PMX1 murine leukemia cells to the intracellular signaling inhibitorsGrowth factor independence is an important step in malignant transformation of hematopoietic cells [25]. To pinpoint mechanisms collaborating with Abl1 deletion in transformation of hematopoietic cells, Abl1 was knocked down in 32Dcl3 cells by CRISPR/Cas9 (using two gRNAs, g1 and g2, see Supplemental Table S2) to obtain 32Dcl3-Abl1ko cells (Fig. 3A). 32Dcl3-Abl1 (Control, V2 vector) and 32Dcl3-Abl1ko cells were challenged to achieve early transformation potential by starving them from growth factors (IL3). Only 32Dcl3-Abl1ko cell populations (1 and 2, derived from g1 and g2, respectively) generated growth factor-independent cells (Fig. 3B). This indicates that the absence of Abl1 may facilitate malignant transformation of hematopoietic cells.
Fig. 3: The impact of ABL1 on response to PI3K inhibitor.
A CRISPR/Cas9-mediated deletion of Abl1 in 32Dcl3-Abl1ko cells. Left panel – Abl1 detected by T7E1 assay. Right panel – Western blot of ABL1 and Actin. B Cell survival curves of 32Dcl3-Abl1 (Control V2 vector) and 32Dcl3-Abl1ko cells following gradual IL-3 withdrawal. Inset - Expression of ABL1 and Actin in two 32Dcl3-Abl1ko clones (1 and 2) growing without IL-3 and in 32Dcl3-Abl1 cells growing in the presence of IL-3 for 2 months after the start of the experiment. C Ingenuity Pathway Analysis (IPA) of the genes deregulated in the 32Dcl3-Abl1ko/IL3- cells using Fragments per Kilobase per Million mapped reads (FPKM) values. Analysis demonstrating top 30 significant canonical pathways. D Venn diagram showing the number of genes which expression is uniquely deregulated in 32Dcl3-Abl1ko cells growing in the absence of IL-3 when compared to 32Dcl3-Abl1ko and 32Dcl3-Abl1 cells growing in the presence of IL-3. E KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways that are altered in the genes at a threshold count of 2 and EASE (the Expression Analysis Systematic Explorer) value of 0.1. Histogram bars represent the fold enrichment score calculated for genes in each pathway. PI3K/AKT pathway is marked by red arrow in panels C and E. F, G Lin-c-Kit+ NUP98-PMX1;Abl1−/−, NUP98-PMX1;Abl1 + /+, AML1-ETO;Abl1−/−, and AML1-ETO;Abl1 + /+ murine bone marrow cells were treated with the indicated concentrations of PI3K inhibitor buparlisib. After 72 h cells were plated in methylcellulose and colonies were counted 7 days later. Results represent mean % ± SD of colonies when compared to untreated controls. Insets: cells were treated with 5 μM buparlisib for 24 h. Total cell lysates were analyzed by Western blot detecting indicated proteins (p = phosphorylated). H Lin-c-Kit+ NUP98-PMX1;Abl1 + /+ cells were treated or not with 2μM ABL1i imatinib + /− the indicated concentrations of buparlisib. After 72 h cells were plated in methylcellulose and colonies were counted 7 days later. Results represent mean % ± SD of colonies when compared to untreated controls.
RNA-seq was performed to compare gene expression in 32Dcl3-Abl1 and 32Dcl3-Abl1ko cells in the presence or absence of IL-3. Pathway analysis of FPKM reads in IL3-independent (32Dcl3-Abl1ko/IL3-) cells showed altered expression of major genes involved in different biological pathways (Supplemental Table S3). Results from the pathway analysis showed that the elements of PI3K-AKT signaling pathway are enriched among the top 30 significant canonical pathways in 32Dcl3-Abl1ko/IL3- cells (Fig. 3C).
Furthermore, to identify the genes and pathways that are uniquely expressed in 32Dcl3-Abl1ko/IL3- cells compared to 32Dcl3-Abl1/IL3 + and 32Dcl3-Abl1ko/IL3 + cells, we performed Venn diagram analysis (Fig. 3D) followed by KEGG pathway analysis (Fig. 3E). RNA-seq analysis detected 291 genes uniquely expressed in 32Dcl3-Abl1ko/IL-3- cells when compared to 32Dcl3-Abl1/IL-3+ cells and 32Dcl3-Abl1ko/IL-3+ cells (Fig. 3D, Supplemental Table S4). Again, PI3K-AKT signaling pathway was significantly altered in 32Dcl3-Abl1ko/IL-3- cells (Fig. 3E).
Since PI3K-AKT pathway was identified in two independent analyses, we tested the sensitivity of AML1-ETO and NUP98-PMX1 –positive Abl1−/− and Abl1 + /+ cells to PI3K inhibitor buparlisib (NVP-BKM120), a selective inhibitor of PI3K p110α/β/δ/γ subunits [26] which reduced phosphorylation of AKT (Fig. 3F, Gleft panels, insets). NUP98-PMX1;Abl1−/− murine leukemia cells were highly sensitive to buparlisib when compared to NUP98-PMX1;Abl1 + /+ counterparts (Fig. 3F, left panel), at the same time ABL1 did not affect the sensitivity of AML1-ETO cells to the inhibitor (Fig. 3G, left panel).
To test if ABL1-positive NUP98-PMX1 leukemia cells may be sensitized to PI3K inhibitor, cells were treated with ABL1 kinase inhibitor imatinib and PI3K inhibitor buparlisib followed by clonogenic assay. The results clearly show that imatinib increased the sensitivity of NUP98-PMX1 leukemia cells to PI3Ki (Fig. 3H).
FPKM and KEGG pathway analyzes suggested potential involvement of mechanisms interacting with PI3K-AKT, including mTOR and RAS-RAF1 [27,28,29] (Fig. 3C–E). mTOR inhibitor rapamycin [30] inhibited phosphorylation of S6 kinase (Supplemental Fig. S3A, B, insets) and was selectively toxic for NUP98-PMX1;Abl1−/− cells when compared to NUP98-PMX1;Abl1 + /+ counterparts (Supplemental Fig. S3A), whereas AML1-ETO -positive Abl1−/− and Abl1 + /+ cells were equally sensitive to the inhibitor (Supplemental Fig. S3B). The presence or absence of ABL1 did not affect the sensitivity of AML1-ETO and NUP98-PMX1 –positive cells to RAF1 inhibitor LY3009120 (data not shown).
In summary, NUP98-PMX1;Abl1−/− cells were highly sensitive to PI3K and mTOR inhibitors when compared to the Abl1 + /+ counterparts, and imatinib sensitized Abl1 + /+ leukemia cells to PI3K inhibitor buparlisib. The resistance of NUP98-PMX1;Abl1 + /+ cells PI3K and mTOR inhibitors might depend on the activation of a pathway redundant to PI3K/AKT/mTOR [31].
ABL1 regulated the sensitivity of AML1-ETO and NUP98-PMX1 -positive cells to DDR inhibitorsSince the role of ABL1 in DDR is well established [32], we tested the sensitivity of AML1-ETO;Abl1−/− and NUP98-PMX1;Abl1−/− Lin-c-Kit+ cells to the inhibitors of DDR and also to DNA damaging agent doxorubicin to identify potential therapeutic vulnerabilities.
ABL1 kinase functionally interacts with DNA-PKcs, ATM and ATR, three major serine/threonine kinases regulating DDR [33, 34]. Therefore, we treated AML1-ETO;Abl1−/− and NUP98-PMX1;Abl1−/− cells and their Abl1 + /+ counterparts with ATM inhibitor (ATMi) KU-60019, ATR inhibitor (ATRi) 504972 and DNA-PKcs inhibitor (DNA-PKi) 260961. Inhibition of these kinases was confirmed by Western blots detecting reduced phosphorylation of their substrates (CHK1, CHK2, H2AX) (Fig. 4A, B, ATMi, ATRi, DNA-PKi insets). AML1-ETO;Abl1−/− cells were modestly more sensitive to all three inhibitors when compared to AML1-ETO;Abl1 + /+ cells (Fig. 4A, ATMi, ATRi, DNA-PKi). Remarkably, NUP98-PMX1;Abl1−/− cells were exceptionally sensitive to ATRi, but displayed similar sensitivity to ATMi and DNA-PKi when compared to NUP98-PMX1;Abl+/+ cells.
Fig. 4: The impact of ABL1 on response to the drugs affecting DDR.
A Lin-c-Kit+ NUP98-PMX1;Abl1−/− and NUP98-PMX1;Abl1 + /+ cells, and B Lin-c-Kit+ AML1-ETO;Abl1−/− and AML1-ETO;Abl1 + /+ cells were treated with the indicated concentrations of doxorubicin, PARPi olaparib, RAD52i 6-hydroxy-DL-dopa, ATMi KU-60019, ATRi 504972 and DNA-PKcsi 260961. After 72 h cells were plated in methylcellulose and colonies were counted 7 days later. Results represent mean % ± SD of colonies when compared to untreated controls (3 independent experiments). Insets: cells were treated with the inhibitors for 24 h. Total cell lysates were analyzed by Western blot detecting indicated proteins (p = phosphorylated). C, D Lin-c-Kit+ NUP98-PMX1;Abl1 + /+ and AML1-ETO;Abl1 + /+ cells were treated or not with 2 μM ABL1i imatinib + /- the indicated concentrations of DNA-PKcsi and ATRi. After 72 h cells were plated in methylcellulose and colonies were counted 7 days later. Results represent mean % ± SD of colonies when compared to untreated controls (3 independent experiments).
In addition, ABL1 kinase phosphorylates and regulates the activity of two DNA repair enzymes, PARP1 and RAD52, inhibition of which triggered synthetic lethality in BRCA1/2-deficient leukemia cells [14, 35,36,37]. We and others reported that AML1-ETO caused “BRCAness” phenotype in leukemia cells [35, 38], but loss of ABL1 expression did not sensitized leukemia cells to PARPi olaparib and RAD52i 6-hydroxy-DL-Dopa (Fig. 4A, B, PARPi and RAD52i). Both inhibitors were validated by detection of elevated levels of DNA double-strand breaks (DSBs marked by γ-H2AX) in the treated cells (Fig. 4A, B, PARPi and RAD52i insets).
Although ABL1 kinase affected survival/apoptosis after genotoxic treatment by regulation of p53 and p73 [39,40,41,42], it did not affect the sensitivity of AML1-ETO and NUP98-PMX1 -positive cells to doxorubicin (Fig. 4A, B, Genotoxic). Genotoxic effect of doxorubicin was confirmed by elevated levels of DSBs marked by γ-H2AX (Fig. 4A, B, Genotoxic, insets).
To test if ABL1-positive NUP98-PMX1 and AML1-ETO leukemia cells may be sensitized to ATRi and DNA-PKi, respectively, cells were treated with ABL1 kinase inhibitor imatinib and 504972 and 260961 compounds followed by clonogenic assay. The results clearly show that imatinib increased the sensitivity of NUP98-PMX1 and AML1-ETO leukemia cells to ATRi and DNA-PKi, respectively (Fig. 4C, D).
留言 (0)