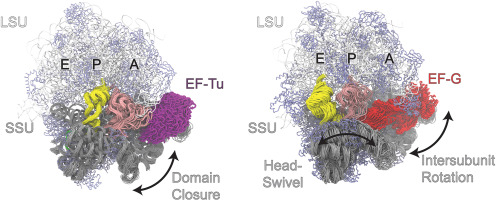
The time- and length-scales accessible to any given type of modeling and simulation technique are limited. Despite consistent advances of modern computing technologies, the need to utilize different simulation models persists, each with different levels of resolution and fidelity as well as varying computational requirements. Multiscale simulations are key to circumventing these limitations, as they facilitate combining information and/or models that capture different spatial or temporal scales. Multiscale frameworks address the contention between access to long- and large-scale dynamics and the computational viability of high-fidelity models. Indeed, multiscale techniques now form the backbone of scientific enquiries in structural biology [1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11∗∗, 12] and almost all other areas of science and engineering [1,4,5,8, 9, 10,12,13].
Multiscale approaches in the field of structural biology encompass a wide range of topics, and the study of complex membrane-protein systems is an important area of investigation and has been used for developing multiscale methods. Developing methods for distilling information from one scale, for example, all atomistic (AA) resolution to coarse-grained (CG) models or vice versa, are ubiquitous [14,15]. Accelerated molecular dynamics (MD) and enhanced sampling methods [16] are also crucial for computational modeling and simulations of complex biological systems. In this perspective, we focus on a rapidly evolving class of techniques for facilitating multiscale simulations for structural biology—those that utilize machine learning (ML).
The past decade has seen ML technologies, in particular, deep learning (DL) [17], creating capability with far-reaching implications in structural biology. DL models are considered universal function approximators [18,19], that is, they can approximate any complex but continuous mapping between inputs and outputs through an appropriately designed neural networks (NNs). This property obviates the need to define such mappings a priori, instead of learning necessary function approximations through vast amounts of data. The tremendous growth in computing—consequential for simulating new data and training DL models—as well as advances in modern, higher-throughput instruments for data capturing (such as X-ray, cryo-EM, and NMR) is enabling DL to play an integral role in contemporary biological applications.
DL techniques are influencing multiscale modeling and simulations in numerous ways [20,21]. For example, DL systems have shown great success as surrogate models [22] as well as in generating spatial structures from sequences of amino acids [23,24] and highly accurate CG force fields for specific biological systems [25,26]. DL is also being used in novel ways for analyzing complex data, for example, capturing membrane lipid fingerprints at different scales [27,28]. ML-based techniques (including DL) are also playing key roles for steering large ensemble simulations [11,29,30].
An important and noteworthy application of DL in structural biology is the technology to accurately predict low-energy protein structures from linear sequences of amino acids. In particular, AlphaFold [23] has outperformed the traditional methods of predicting protein structures [24,31,32]. Despite the impressive success and potential of AlphaFold [33], some challenges remain [34], such as predicting multi-protein components, metal ions, cofactors, and other ligands. To overcome these challenges, there are various efforts underway to capture protein interactions, such AlphaFold Multimer [35], RoseTTAFold [36], and ESMFold [37]. Although such methods facilitate working across “scales” (i.e. primary to tertiary structures), they are not considered multiscale techniques in the usual sense. As such, although they offer substantial promise for structural biology, they will not be discussed further in this review.
Traditional multiscale approaches have been classified as serial or parallel [38]. Serial or sequential multiscale methods resolve or collapse degrees of freedom across scales a priori and utilize information from the finer scale to parameterize coarser scales and/or sampling at coarser scales to instantiate the finer scale. Parallel or concurrent multiscale methods, on the other hand, are coupled and perform cross-scale information exchange inside the running multiscale simulations, where specific regions or molecules of interest are often represented at a finer scale and coupled, through specific annealing regions or using cross–scale parameters, to a coarser scale used for the bulk environment. Recent works on ML-driven ensemble-based coupled multiscale simulations [11,30] leverage the simplicity of serial multiscale methods while coupling in parallel the coarser macro model to continue improvements from concurrently running finer-scale simulations.
In this paper, we focus on two broad applications of ML for facilitating multiscale simulations in structural biology. The first is in the context of scale bridging. Several ML techniques have been proposed to transform data from one scale to another, for example, coarse-graining of AA configurations [39, 40, 41] , as well as backmapping approaches, for example, from CG to AA [42, 43, 44∗]. The second class of techniques focuses on sampling and control of simulations using ML, for example, to identify when and where to promote configurations to finer scales [11] or to stop simulations that explore uninteresting regions of phase space [45]. Both classes of techniques are key to enabling large multiscale simulations, especially when leveraging modern computing resources.
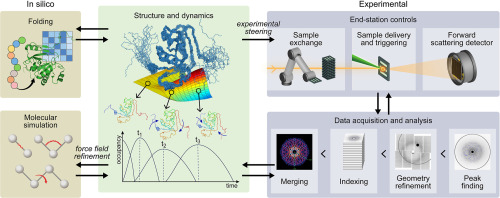
The unprecedented scale of modern computing resources offers exciting opportunities for scientific applications; accompanied are many challenges in making efficient use of these software and hardware resources. The high-performance computing (HPC) community is moving away from large and monolithic codes to sophisticated workflows [46, 47, 48, 49] that create massive simulation ensembles. Traditional metrics for scaling, such as strong and weak scaling, are getting replaced by the need for simultaneous utilization of heterogenous resources, tailored to the needs of multiscale. ML-based techniques have demonstrated immense value in facilitating such a vision through the automated/semi-automated frameworks that rely on ML to generate targeted or exploratory ensembles of multiscale simulations (Figure 1) [11,30,50, 51∗∗, 52∗, 53, 54]. Multiscale frameworks powered by ML are paving the way for a new revolutionary approach for studying scientific phenomena. Such methods are likely going to be the centerpiece of computational sciences in the exascale era.









留言 (0)