Specimens of a clinical nature
Histologically confirmed NSCLC tissue and serum samples (10 with metastasis and 10 without metastasis) were obtained from 20 patients who underwent surgery in Suzhou Municipal Hospital. 20 healthy serum samples matched the gender and age of the above patients were obtained from the Department of Respiratory Medicine, Suzhou Municipal Hospital. All clinical specimens were stored at − 80 °C after collection and frozen in liquid nitrogen. This study was approved by the Ethics Committee of Suzhou Hospital Affiliated to Nanjing Medical University, and all participants signed written consent.
Cell culture
Human normal bronchial epithelial cells (16HBE) and NSCLC cell lines PC9, A549 and H1299 were purchased from the American Type Culture Collection (ATCC). Cells were cultured in RPMI 1640 medium supplemented with 1% penicillin/streptomycin, 10% fetal bovine serum (FBS) in a 37 °C and 5% CO2 incubator. All media and reagents were purchased from Gibco, USA.
Exosomes isolation and characterization
Exosomes were extracted from NSCLC patient serum and NSCLC culture medium by ultracentrifugation. After the cells reached about 60% confluency, the medium was changed to RPMI 1640 medium containing 10% exosome-depleted FBS, and the cell supernatant was collected 2 days later and centrifuged at 500 g/16,800 g, respectively, at 4 °C. 10/30 min. The supernatant was separated, passed through a 0.22 um filter (Millipore) and then ultracentrifuged at 110,000 g for 70 min at 4 °C. After washing with phosphate buffered saline (PBS), ultracentrifuge at 110,000 g for 70 min at 4 °C and resuspend in PBS. Exocrine quantification by BCA protein detection kit (KeyGEN BioTECH). Exosome morphology was observed using a Tecnai T20 transmission electron microscope (TEM) from FEI, and the size and number of exosomes were determined by Nano Sight NS 300 system (Nano Sight Technology, Malvern, UK). Fluorescent labeling of exosomes with Sigma PKH67 membrane dye (green). Stained exosomes were washed in 10 ml PBS, then collected by ultracentrifugation and resuspended in PBS. For cellular uptake of exosomes, 2 µg of exosomes were incubated with 2 × 105 recipient cells for 48 h.
Quantitative real time‐polymerase chain reaction (qRT‐PCR)
Total RNA was first extracted from exosomes and cells using Trizol reagent (Invitrogen). Then cDNA for miRNA was synthesized using the miDE TECT A Track RT kit (RiboBio, China), while lncRNA and mRNA, complementary cDNA was obtained using a reverse transcription system (Promega, Madison, WI, USA). lncRNA, mRNA and miRNA expression levels were measured in triplicate samples by using SYBR Green PCR Master Mix (Applied Bio systems, Carlsbad, CA) on an ABI 7900 PCR system (Applied Bio systems) according to the manufacturer’s instructions. U6 and ACTB was selected as a normalization control for miRNA, lncRNA and mRNA sum, respectively.
FISH
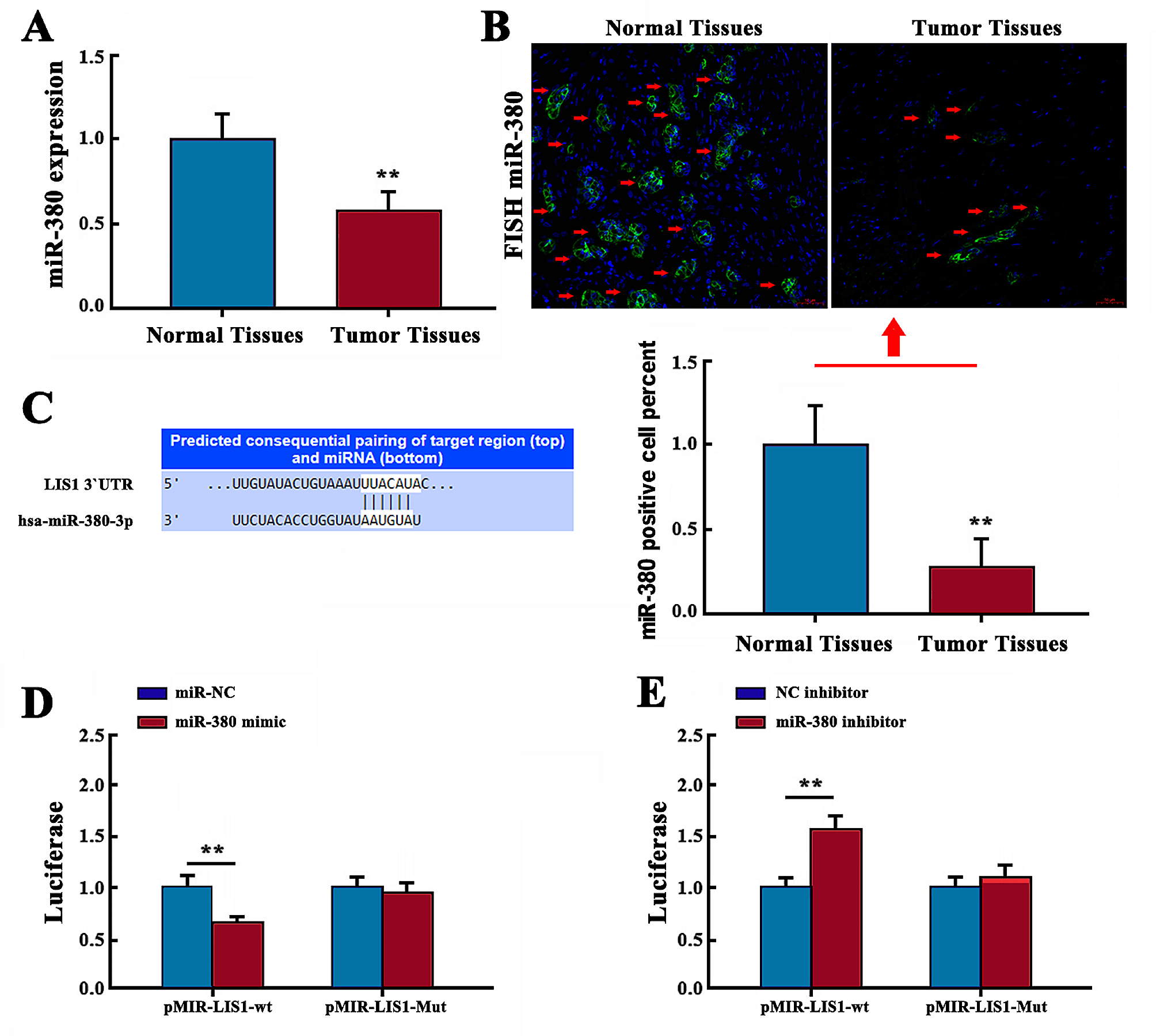
The relative expression of MFI2-AS1 in NSCLC tissues was examined using RNA fluorescence in situ hybridization (FISH) staining. Paraffin sections were dewaxed and rehydrated with ethanol. Paraffin sections were digested with proteinase K, fixed in 4% paraformaldehyde, hybridized overnight with a digoxigenin-labeled MFI2-AS1 synthetic oligonucleotide probe, followed by anti-digoxigenin-labeled peroxidase (anti-DIG Horseradish peroxidase [HRP]) (Roche, Mannheim, Germany) was incubated for 2 h. Subsequently, the paraffin sections were washed with water for 10 min and counterstained with hematoxylin. Images were observed and taken by using a fluorescence microscope (Olympus).
Plasmid construction and transfection
We construct plasmids to knock down or increase the expression of cellular genes. PC9/A549 cells were plated at a cell density of 70% dish confluence for transfection. Plasmids transfected with Lipofectamine 2000 (Invitrogen, Carlsbad, CA). Plasmids for MFI2-AS1, NFAT5, miR-107 inhibitor or small interfering RNA (si-RNA) of MFI2-AS1 and NFAT5 were constructed by Genechem (Shanghai, China).
Dual luciferase reporter gene assay
The pmirGLO Dual-Luciferase miRNATarget expression vector was purchased from GenePharma (Shanghai, China), and the luciferase reporter plasmid was inserted into the wild-type WT-MFI2-AS1 and Mut-MFI2-AS1 3'UTR sequences. Firefly luciferase was used as the primary reporter gene to regulate mRNA expression, and Renilla luciferase was used as a normalization control. Dual-luciferase reporter gene assay system (Promega) was used 48 h after transfection. The mean luciferase intensity was normalized to Renilla luciferase. Data are shown as mean ± SD and each experiment was performed three times.
Western blot
Cells were harvested after 24 h of co-culture of exosomes with cells. After RIPA cleavage, total protein was extracted and assayed by BCA method. Quantitative denatured proteins (20 μg protein per lane) were separated on 10% SDS-PAGE and transferred to protein electrophoresis membranes, which were blocked with fast sealing solution. Follow the procedure for the first and second incubations. The expression of proteins is represented by grayscale values. List of primary antibodies: CD63 (ab134045, Abcam), Calnexin (ab133615, bacam), Tsg101 (ab125011, Abcam), NFAT5 (ab3446, Abcam), aAKT/p-AKT (CST, #4060, GAPDH (ab8245, Abcam).
CCK8
We used Cell Counting Kit-8 (CCK8) to observe cell proliferation. The co-cultured cells were seeded into 96-well plates at 1000 cells/well, with 3 replicate wells per group. 2 h before observation, 10 μL of CCK8 reagent was added to each well for treatment. Finally, use absorbance at 450 nm in the dark to determine cell proliferation viability. Each sample were performed in three times.
Tube formation
250 μL of Matrigel (BD Bioscience, USA) was added to a 48-well plate and resuspended at approximately 2 × 104 cells per well. Tube formation was observed with an inverted microscope (Olympus Corporation, Tokyo, Japan) after 8–12 h of incubation. Quantify the branching of the tube structure according to the manufacturer's instructions after taking the images. Use Image J to measure and calculate the total length of tube formation in each well. Each sample was replicated three times.
Wound-healing assay
Cells were resuspended and plated in serum-free medium and replated in 6-well plates to grow to 100% confluency. After scraping the cell monolayer vertically with a sterile pipette tip, wash the cells 3 times with 1% PBS. Wound fusion was observed under an inverted microscope (Olympus Corporation, Tokyo, Japan) and photographed at 0 and 36 h. Each sample was replicated three times.
Transwell
Transwell migration assays used 8.0 μm Transwell permeable supports (Corning, New York, USA). Briefly, 4 × 104 cells in 400 μL of serum-free medium were added to the upper chamber. Add 600 µL of medium containing 10% FBS to the lower chamber. The chambers were incubated in 5% CO2 at 37 °C for 24 h and fixed with paraformaldehyde for 30 min. After staining with 0.4% crystal violet for 15 min, cells were photographed and counted in 5 random fields. Each sample was replicated three times.
RIP
We transfected HUVECs with pcDNA3.1-MFI2-AS1 or control for 48 h, then resuspended cells in radioimmunoprecipitation assay (RIPA) buffer and incubated for 30 min. After centrifugation, human anti-Ago2 antibody (Proteintech, USA) was added to the lysates and incubated for 4 h, selecting normal mouse immunoglobulin G as a negative control. We then resuspended the beads in RIPA buffer and treated the samples with proteinase K for 45 min in a 45 °C incubator. Trizol extracted immunoprecipitated RNA samples were tested by quantitative real-time PCR analysis.
Animal experiment
5 × 106 A549 cells were injected subcutaneously into the armpits of 4-week-old female nude mice. 20 mg of exosomes were injected into the right axillary tumor center of mice every 3 days. After 30 days, the mice were sacrificed, the subcutaneous tumors were removed, and the volume and weight were recorded. Afterwards, tumors were excised for immunohistochemical staining for CD34. All animal experiments were approved by the Animal Research Ethics Committee of Nanjing Medical University.
Statistical analysis
SPSS19 Statistics software was used for statistical data analysis. Data are presented as mean ± standard deviation (SD) and each experiment was performed three times.
Student’s t-test or one-way analysis of variance (ANOVA) was used to compare statistics between two or more groups. P < 0.05 was considered statistically significant.

留言 (0)