
記住我


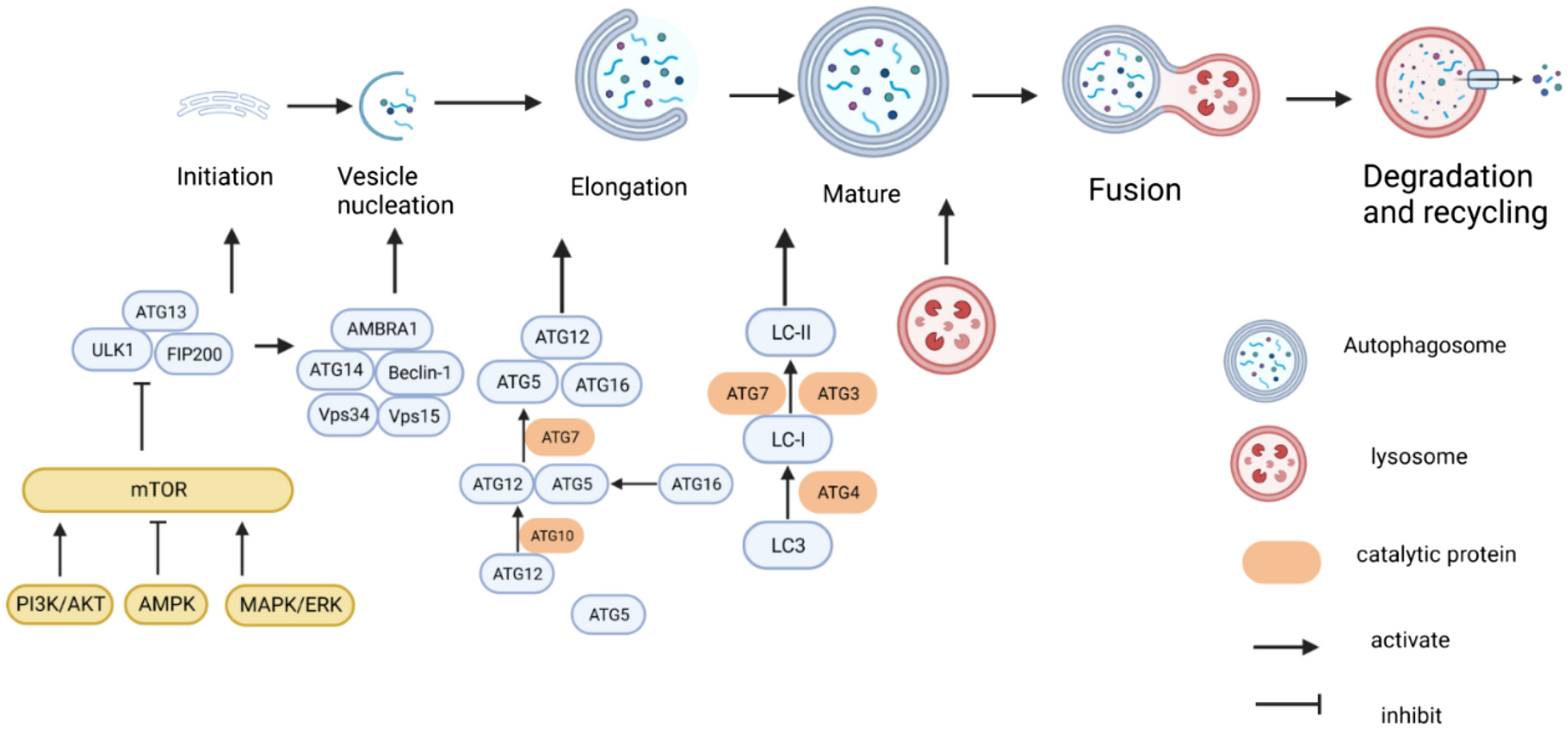

Because the view that remote ischemic preconditioning (RIPC) protects from SCIRI is still controversia [24, 40], we first assessed if RIPC protects mice from spinal cord I/R using the strategy shown on Fig. 1A. The Basso mouse scale (BMS) analysis was performed until 3 days after ischemic. As shown in Fig. 1B, RIPC did not adversely affect the motor functions of uninjured mice, and the recovery of lower limb function within 3 days after ischemia in the RIPC + I/R group was better than in the I/R group. To assess post-ischemic hind limb balance, we performed footprint analysis on the third day after ischemia. As shown in Fig. 1C, D, mice in the RIPC + I/R group exhibited better coordination recovery, as evidenced by longer stride lengths and shorter stride width compared to the I/R group. Furthermore, we found that in the RIPC + I/R group, RIPC led to higher motor-evoked potentials (MEPs) amplitudes when compared with mice in the I/R group (Fig. 1E–F). Analysis of neuron degeneration using Nissl staining revealed that RIPC did not significantly affect Nissl body morphology and number, and that I/R was associated with weak of Nissl staining and reduced Nissl body numbers in the ventral lateral anterior horn motor neurons of the mouse lumbar spinal cord (Fig. 1G, H). This change was markedly reversed by performing RIPC before I/R. Because spinal cord I/R triggers increased neuronal apoptosis, we used TUNEL analysis to assess the rate of neuronal apoptosis. This analysis found that after I/R, the number of neurons (in green) reduced while the number of TUNEL-positive cells (red) increased, and that this change was partially reversed by RIPC (Fig. 1I, J). Redox imbalance is a key pathogenic factor in I/R injury. To determine if oxidative stress was inhibited by RIPC, we measured the levels of oxidative indexes malondialdehyde (MDA) and antioxidant indexes, including superoxide dismutase (MnSOD), catalase (CAT) and reduced-glutathione (GSH). This analysis showed that I/R reduced the activity of MnSOD and CAT, and the content of GSH in spinal cord while enhancing MDA expression (Fig. 1K). However, the above indexes were partially reversed in the RIPC + I/R group. These results indicated that RIPC-mediated inhibition of oxidative stress after spinal cord I/R is neuroprotective in mice.
Fig. 1
RIPC protects against SCIRI in mice. A Schematic diagram of the experimental design. B BMS scores at different time points post-injury in mice (n = 5/group). C Representative footprint images of mice on the third day following injury. Blue: frontpaw print; red: hindpaw print. D Quantification of the analysis of the footprint in each group (n = 5/group). E Representative images of MEP for assessing the electrophysiology of mice on the third day after injury. F Quantification of the peak-to-peak MEP amplitudes in each group (n = 5/group). G Representative images of Nissl staining of neurons in the anterior horn of the spinal cord. Scale bar = 100 μm. H Quantification of the number of integrated Nissl bodies per section (n = 5/group). I Representative images of TUNEL-positive apoptotic cells (in red) in spinal cord sections on day 3 post-injury. Neuron was stained with NeuN (in green) and nuclear stained with DAPI (in blue). Scale bar = 100 μm. J Quantification of the number of apoptotic cells in each group (n = 5/group). K MDA, MnSOD, CAT, and GSH were measured to reflected the level of oxidative stress in each group (n = 5/group). Statistical analysis: mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001
SIRT3 is essential for RIPC-mediated neuroprotectionTo investigate the role of SIRT3 in RIPC-mediated neuroprotection, we first examined SIRT3 expression at various timepoints after SCIRI. Western blot revealed that SIRT3 levels fell significantly 6 h after reperfusion and remained low for 24 h (Fig. 2A, B), indicating that SIRT3 downregulation may contribute to the deterioration of neurological function in mice after SCIRI. Next, we investigated whether the neuroprotective effects of RIPC was mediated by SIRT3, western blotting results showed that RIPC enhanced SIRT3 expression and partially rescued SIRT3 downregulation caused by I/R (Fig. 2C, D). This conclusion was further confirmed by immunofluorescence staining of SIRT3 in neurons (Fig. 2E, F). To further determine if SIRT3 was necessary for RIPC-mediated neuroprotection, we generated Sirt3-KO mice as previously reported [41] and subjected them to RIPC and I/R injury. Genotyping of KO mice was confirmed using DNA that was isolated from tail clips (Additional file 1: Fig. S1). Analysis of mouse neurological recovery using BMS scores, footprint analysis, and MEPs revealed that, when compared with WT mice (Fig. 1B–F), RIPC did not enhance functional recovery in KO mice (Fig. 2G–K). Moreover, Nissl staining showed that RIPC did not reverse I/R-induced loss of Nissl bodies and morphological changes in KO mice (Fig. 2L, M). The number of TUNEL-positive cells in RIPC + I/R group KO mice was similar to that observed in I/R group (Fig. 2N, O). These results indicated that SIRT3 was essential for RIPC-mediated neuroprotection. To determine if SIRT3 contributes to RIPC-mediated neuroprotection by regulating the activity of antioxidant enzymes, we assessed the activity of SOD, and CAT, as well as GSH content. Results showed that RIPC-induced activation of SOD, CAT, and GSH in WT mice were markedly suppressed in KO mice (Fig. 2P). Moreover, I/R induced a more significant increase in MDA content in KO mice when compared to WT mice, and this effect was not significantly ameliorated by RIPC. Together, these results indicated that SIRT3 plays a key role in RIPC-mediated tolerance to I/R and resistance to oxidation in vivo.
Fig. 2
RIPC upregulates SIRT3 expression and the neuroprotective effect of RIPC is reversed in KO mice. A, B Western blot was used to detect the expression of SIRT3 at different time points after spinal cord I/R. Relative expression of SIRT3 was normalized to the level of control. N = 5/group, *p < 0.05, **p < 0.01, compared to the former timepoint. C, D Western blot was used to detect the expression of SIRT3 in different groups. Relative expression of SIRT3 was normalized to the level of the sham group (n = 5/group). E, F Immunofluorescence was used to detect the expression of SIRT3 (green) in neurons (red) in different groups. Nuclear was stained with DAPI (in blue). Scale bar = 100 μm. Relative fluorescence intensity was normalized to the level of the sham group (n = 5/group). G The KO mice were functionally scored up to 3d postinjury using BMS (n = 5/group). H, I Representative footprint images of KO mice on the third-day post-injury and quantification of the stride length and width. Blue: frontpaw print; red: hindpaw print (n = 5/group). J, K MEP analysis was used for electrophysiological assessment at day3 postinjury and quantification of the peak-to-peak MEP amplitudes in KO mice (n = 5/group). L, M Representative images of Nissl staining of neurons in the anterior horn of the spinal cord and quantification of the number of integrated Nissl bodies per section from KO mice (n = 5/group). Scale bar = 100 μm. N, O Representative images of TUNEL-positive apoptotic cells (in red) in spinal cord sections and quantification of the number of apoptotic cells in each group from KO mice at day 3 postinjury (n = 5/group). Neuron was stained with NeuN (in green) and Nuclear stained with DAPI (in blue). Scale bar = 100 μm. P MnSOD, GSH, CAT, and MDA were measured to reflect the level of oxidative stress in each group of KO mice (n = 5/group). Statistical analysis: mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001
RIPC regulates SIRT3 expression via NMDARAlthough we found that RICP protects neurons from SCIRI by modulating SIRT3, it is not clear how RIPC induces SIRT3 expression. Previous finding have shown that RIPC enhances glutamic levels in the spinal ventral horn and sub-lethally activates N-methyl-D-aspartate receptor (NMDAR) [34]. Several studies have shown that NMDAR is involved in the protective effects of IPC [42, 43]. However, it is not clear if NMDAR participates in RIPC-mediated neuroprotection by modulating SIRT3. To determine the relationship between NMDAR and SIRT3, immunofluorescence was performed to assess NMDAR expression in spinal cord neurons. As shown in Fig. 3A, B, NMDAR levels were elevated by RIPC and further enhanced after I/R. However, performing RIPC before I/R attenuated SCIRI-induced NMDAR overactivation (Fig. 3A, B). Similar effects were observed through western blotting (Fig. 3C). Next, to inhibit the NMDA receptor, we intravenously treated preconditioned mice with NMDA receptor inhibitor, dizocilpine (MK-801, 1 mg/kg), 60 min before remote ischemic preconditioning and evaluated SIRT3 expression. Immunofluorescence and western blot revealed that MK-801 suppressed RIPC-induced SIRT3 upregulation (Fig. 3D–F), indicating that RIPC may regulate SIRT3 expression through glutamate and the glutamate receptor, NMDAR.
Fig. 3
RIPC fails to upregulate SIRT3 after NMDAR inhibition. A, B Immunofluorescence was used to detect the expression of NMDAR2B (in green) in neurons (in red) in different groups (n = 5/group). Nuclear was stained with DAPI (in blue). Scale bar = 100 μm. C Western blotting was used to detect the expression of NMDAR2B in different groups (n = 5/group). D, E The expression of SIRT3 (in green) in the spinal cord neurons (in red) was detected by immunofluorescence after administration of NMDR2B inhibitor MK-801 (n = 5/group). Scale bar = 100 μm. F The expression of SIRT3 in the spinal cord was detected by Western blotting after administration of MK-801 (n = 5/group). G Western blotting was used to detect the expression of SIRT3 at different time points after OGD/R (n = 5/group). H The expression of SIRT3 in neurons was detected by Western blotting after different treatment conditions and NMDA partially reversed the down-regulation of SIRT3 in neurons after OGD/R (n = 3/group). I The expression of SIRT3 in neurons was detected by Western blotting after different treatment conditions and sublethal glutamate partially reversed the down-regulation of SIRT3 in neurons after OGD/R (n = 5/group). Statistical analysis: mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001
SIRT3 deficiency exacerbates OGDR-induced neuronal injury and suppresses NMDAR agonists-mediated protection in vitroTo further investigate the relationship between NMDAR and SIRT3, we extracted primary cortical neurons from WT and KO embryonic mice, and performed oxygen–glucose deprivation and reoxyglucose (OGD/R) to simulate ischemia–reperfusion injury. The purity of the neurons was identified by co-staining MAP2 and NeuN, and neurons with purity greater than 90% were used in subsequent experiments (Additional file 1: Fig. S2). Next, we examined SIRT3 levels in response to OGD/R. Similar to the in vivo results, neuronal SIRT3 expression reduced with increasing duration of OGD/R treatment (Fig. 3G). Neuronal Stimulation using the NMDAR agonist, NMDA (100 μm) or sublethal glutamate (10 μm) for 6 h markedly elevated SIRT3 expression and reversed OGD/R-induced SIRT3 downregulation (Fig. 3I–H). These results confirmed that NMDAR may control neuronal ischemic tolerance by modulating SIRT3 expression. To determine if NMDA protects neurons from OGD/R in a SIRT3-dependent manner, neurons obtained from WT and KO embryonic mice were preconditioned respectively with NMDA for 6 h followed by OGD/R. As shown in Fig. 4A, B, bright field images revealed that NMDA treatment alone had no significant effect on the morphology and number of WT or KO mice derived neurons. However, KO-derived neurons were more susceptible to OGD/R stimulation and the resulting damage could not be reversed by NMDA preconditioning. In contrast, NMDA pretreatment improved the morphology and abundance of WT-derived neurons after OGD/R. Similarly, SIRT3 deficiency led to more toxic LDH release from neurons, which could not be reversed by NMDA (Fig. 4C). Flow cytometry showed that NMDA pretreatment suppressed apoptosis of WT-derived neurons but not in KO-derived neurons (Fig. 4D, E). In addition, NMDA pretreatment enhanced the activities of the antioxidant enzymes MnSOD and CAT and increased the level of GSH in WT-derived, but had no similar effect on KO-derived neurons (Fig. 4F). Flow cytometry was used to detected ROS generation. The results showed that pretreatment with NMDA suppressed ROS generation in WT-derived neurons after OGD/R, but had no significant effect on ROS production in KO-derived neurons (Fig. 4G, H). Together, these results indicate that SIRT3 plays a key role in NMDAR-mediated ischemic tolerance in vitro.
Fig. 4
NMDA exerts a neuroprotective effect similar to that of RIPC in vitro. A Representative brightfield image showing morphologic changes of primary neurons in WT or KO-derived neurons after NMDA pretreatment. Scale bar = 50 μm. B Quantification of surviving neurons in figure A (n = 3/group). C The percentage of released LDH from OGD/R-treated KO or WT- derived neurons was assessed to determine the neuronal injury in the presence or absence of NMDA (n = 3/group). D Flow cytometry was used to detect the apoptosis of WT or KO-derived neurons pretreated with or without NMDA by PI/Annexin V double labeling. E Quantification of apoptotic cells (Annexin V-positive) in panel C (n = 3/group). F Relative activity of MnSOD, CAT, and GSH content was detected in neurons. Relative activity or content was normalized to the level of WT-derived neurons treated with OGD/R (n = 3/group). G Analysis of ROS production by flow cytometry in KO (left panel) and WT (right panel) derived neurons pretreated with or without NMDA. H Quantification of ROS production in neurons of indicated groups (n = 3/group). Relative fluorescence intensity was normalized to the level of WT neurons that were treated with OGD/R. Statistical analysis: mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001
NMDAR regulates SIRT3 expression via the AMPK/PGC-1α signaling pathwayTo determine the mechanism by which NMDAR modulates SIRT3 expression, we examined the AMPK/PGC-1α signaling pathway, which is reported to act upstream mediator of SIRT3. As shown in Fig. 5A, B, NMDA increased AMPK phosphorylation and PGC-1α as well as SIRT3 expression in a time-dependently manner in neurons. In vivo, RIPC also upregulated the levels of p‐AMPK and PGC-1α in the spinal cord, and reversed p‐AMPK and PGC-1α downregulation induced by I/R (Fig. 5C, D). To further confirm that AMPK is the upstream mediator of RIPC regulating SIRT3 expression, an AMPK inhibitor, compound C and shRNA were used to treated neurons. As shown in Fig. 5E, F, NMDA-induced upregulation of SIRT3 and PGC-1α was significantly suppressed after AMPK inhibition. To identified if AMPK is also involved in SIRT3-dependent neuronal survival and mitochondrial homeostasis, we also assessed the effects of NMDA on neuronal apoptosis and oxidative stress after AMPK inhibition. As shown in Fig. 5G–I, compound C did not affect neuronal viability under normal conditions, but significantly reversed the inhibitory effect of NMDA on OGD/R-induced apoptosis and LDH release. The activity of antioxidant enzymes and the content of GSH and ROS were also measured after AMPK inhibition. As shown in Fig. 5J, AMPK inhibition using compound C after NMDA + OGD/R treatment suppressed the activities of MnSOD and CAT as well as the levels of GSH, while enhancing ROS production (Fig. 5K, L). Together, these results suggest that NMDAR/AMPK/PGC-1α signaling promotes mitochondrial homeostasis and neuronal survival by upregulating SIRT3.
Fig. 5
SIRT3 expression is regulated by the classical AMPK/PGC-1α signaling pathway and inhibition of AMPK attenuates the NMDA-induced neuroprotective effect. A, B Altered protein expression of NMDAR2B, T-AMPK, p-AMPK, PGC-1α and SIRT3 in neurons treated with NMDA was detected by Western blotting at different time points. Relative expression was normalized to the level of the control (n = 3/group). C, D Western blotting was used to detect the expression of T-AMPK, p-AMPK, and PGC-1α after sham or I/R treatment with or without RIPC. Relative expression was normalized to the level of the sham group (n = 5/group). E, F In response to NMDA, altered protein expression levels of p-AMPK, PGC-1α and SIRT3 in neurons when blocking AMPK using AMPK shRNA and compound C (an AMPK inhibitor) were detected using Western blotting. Relative expression was normalized to the level of control (n = 3/group). G Flow cytometry was used to detect the effect of compound C pretreatment on apoptosis of NMDA-OGD/R treated neurons (n = 3/group). H LDH was detected to assess neuronal damage after pretreatment with compound C in NMDA-OGD/R treated neurons (n = 3/group). I The relative activity of MnSOD, CAT, and GSH content was measured after pretreatment with compound C in NMDA-OGD/R treated neurons (n = 3/group). J, K The production of ROS was measured by flow cytometry after pretreatment with compound C in NMDA-OGD/R treated neurons (n = 3/group). Statistical analysis: mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001
Biphasic effects of different Ca2+ concentrations on AMPK activationAs mentioned above, RIPC increased SIRT3 expression through activation of NMDAR. However, after I/R, NMDAR was further activated, but the level of SIRT3 was no longer increased. This observation suggested a biphasic effect of NMDAR on SIRT3 regulation. In vitro, neurons were treated with sublethal (10 μM) and lethal concentrations (100 μM) of glutamate to mimic NMDAR activation by RIPC and I/R in vivo respectively. Neuronal apoptosis and LDH release were detected. The results showed that sublethal glutamate had no effect on neuronal viability, but lethal glutamate resulted in significant neuronal apoptosis and LDH release (Fig. 6A, B). Western blot revealed that sublethal concentrations activated the AMPK/PGC-1/SIRT3 pathway in a time-dependent manner, while lethal glutamate levels markedly inhibited it (Fig. 6C, D). This suggests that various NMDAR activation levels may determine neuronal fate by modulating the AMPK/SIRT3 signaling.
Fig. 6
Toxic concentrations of glutamate inhibits AMPK activation through PP4 activation. A Flow cytometry was used to detect the apoptosis of neurons stimulated with different concentrations of glutamate. Apoptotic neurons were defined as Annexin V-positive cells (n = 3/group). B LDH was detected to assess neuronal damage after stimulation with different concentrations of glutamate (n = 3/group). C, D Altered protein expression levels of NMDAR2B, T-AMPK, p-AMPK, PGC-1α and SIRT3 in neurons treated with different concentrations of glutamate were detected by Western blotting at different time points (n = 3/group). E Flow cytometry was used to detect intracellular Ca2+ concentration in neurons stimulated with different concentrations of glutamate. Relative intensity was normalized to the level of control (n = 3/group). F, G Western blotting was used to detect the expression of T-AMPK, p-AMPK, PGC-1α and SIRT3 after treatment with intracellular calcium chelator BAPTA in 100Glu-treated neurons. Relative expression was normalized to the level of control (n = 3/group). H, I The expression of protein phosphatase-4 was detected using western blotting in neurons after treatment with 100Glu at different time points. Relative expression was normalized to the level of control. * p < 0.05, **p < 0.01, compared to the former timepoint (n = 3/group). J Western blotting was used to detect the expression of T-AMPK, p-AMPK, PGC-1α and SIRT3 after treatment with the PP4 inhibitor cantharidin in 100Glu-treated neurons. Relative expression was normalized to the level of control (n = 3/group). Statistical analysis: mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001
The activation of NMDAR by ligands triggers inward calcium flow. It has been reported that transient Ca2+ level elevation contributed to AMPK activation, while sustained high Ca2+ levels inhibited AMPK activation. Therefore, we speculated that the above difference may be due to different intracellular Ca2+ levels in response to distinct stimuli. To test this possibility, we used Fluo-4, a fluorescent probe, to assess changes in intracellular Ca2+ levels under various stimuli using flow cytometry. As shown in Fig. 6E, 100 μM glutamate caused a markedly higher inward flow of intracellular calcium when compared with 10 μM glutamate. To elucidate the relationship between intracellular Ca2+ concentration and AMPK activation, neurons were treated with the intracellular calcium chelator BAPTA-AM (10 μM) [44] before lethal glutamate administration. Western blot revealed that pretreatment with BAPTA-AM restored AMPK activation as well as the expression of PGC-1α and SIRT3 (Fig. 6F). This observation highlighted the biphasic nature of Ca2+ concentration in AMPK phosphorylation. Calcium/calmodulin-dependent protein kinase β (CaMKKβ) is a serine/threonine-protein kinase belonging to the Ca2+/calmodulin-dependent protein kinase subfamily [45], and AMPK-α is a key CaMKKβ target [46]. Thus, we hypothesized that toxic concentrations of glutamate inhibited AMPK activation by inhibiting CaMKKβ autophosphorylation. However, western blot analysis revealed that treating neurons with toxic glutamate concentrations (100 μM) did not inhibit CaMKKβ activation but instead, enhanced CaMKKβ phosphorylation when compared with 10Glu glutamate (Additional file 1: Fig. S3). Indicating that AMPK activity was suppressed via CaMKKβ-independent mechanism. Because Ca2+ levels may enhance AMPK dephosphorylation by protein phosphatase-4 (PP4) [44], we evaluated neuronal PP4 protein levels after treatment with lethal glutamate concentration (100 μM). Western blot analysis revealed that the lethal glutamate concentration elevated neuronal PP4 protein levels (Fig. 6H, I), indicating a potential link between PP4 expression levels and AMPK activity. Next, we pretreated neurons with the PP4 inhibitor, cantharidin (50 μM), before stimulating them with the lethal glutamate concentration. Western blot showed that cantharidin restored AMPK phosphorylation as well as the expression of PGC-1α and SIRT3 (Fig. 6J), which is consistent with the calcium chelators (Fig. 6F). These findings indicate that NMDAR regulates AMPK activation via inward Ca2+ flow and that transient Ca2+ elevation contributes to AMPK activation, while sustained high Ca2+ levels inhibit AMPK activation by upregulating PP4 expression in neurons.
Honokiol, a SIRT3 agonist, protects neurons from OGD/R-induced oxidative stress damageDespite the considerable potential of RIPC in resistance to SCIRI, we face the challenge of what else can be done after SCIRI. Enhancing neuronal SIRT3 expression before or after injury in order to improve resistance to external stress is a promising direction. Honokiol (HKL, Fig. 7A), a biphenolic compound obtained from the bark of magnolia trees, has been reported to have antioxidative and neuroprotective properties. Moreover, HKL promotes SIRT3 activity by enhancing SIRT3 expression as well as directly binding to SIRT3 and enhancing its deacetylase activity [47, 48]. Thus, HKL may have significant therapeutic potential against SCIRI. To test this possibility, we first assessed the effects of different HKL concentrations on neuronal viability and SIRT3 expression. CCK8 analysis showed that neurons treated with HKL at 10 μM exhibited maximum viability in response to OGD/R, and had no obvious toxic effect on normal neurons (Fig. 7B). Consistently, western blot also revealed that neuronal SIRT3 expression was highest upon treatment with 10 μM HKL in vitro (Fig. 7C, D). Therefore, 10 μM HKL was used in subsequent experiments. Next, we performed oxy-glucose deprivation and then added HKL to the normal neuronal media after re-oxy-glucose onset and maintained for 12 h. Brightfield microscopic examination revealed that HKL partially rescued OGDR-induced neuronal morphological changes and loss (Fig. 7E, F). HKL-mediated resistance to OGD/R was further confirmed by measuring the level of LDH release into the neuronal medium (Fig. 7G). Flow cytometry revealed that HKL suppressed OGD/R-induced neuronal apoptosis (Fig. 7H, I). Notably, we observed that the neuroprotective effects of HKL were markedly attenuated in Sirt3−/−-derived neurons (Fig. 7E–I). Moreover, HKL enhanced the activity of the antioxidant enzymes MnSOD and CAT, increased the GSH level (Fig. 7I), and inhibited OGD/R-induced mitochondrial ROS production in the presence of SIRT3 (Fig. 7J–L). However, this antioxidant effect of HKL was markedly attenuated in Sirt3−/−-derived neurons (Fig. 7J–L). Importantly, we found that in OGD/R treated neurons, HKL upregulated SIRT3 expression by activating AMPK-PGC-1α signaling and suppressed NMDAR overaction (Fig. 7M, N). These results indicate that HKL protects neurons from OGD/R-induced oxidative stress by at least in part, modulating NMDAR/AMPK/PGC-1α/SIRT3 signaling.
Fig. 7
HKL attenuates OGD/R-induced damage by preventing neuronal apoptosis and oxidative stress via the NMDAR/AMPK/PGC-1α/SIRT3 pathway in neurons. A The chemical structure of HKL. B The CCK8 kit was used to detect the viability of neurons that were treated with different concentrations of HKL with or without OGD/R (n = 6/group). * p < 0.05, compared to the former group. C, D Immunoblot images of SIRT3 in neurons treated with different concentrations of HKL. Relative expression was normalized to the level of the control treatment (n = 3/group). *p < 0.05, **p < 0.01, compared to the former group. E Representative brightfield images showing morphologic changes of OGD/R-treated primary neurons in the presence or absence of HKL. Scale bar = 50 μm. F Quantification of surviving neurons in figure E (n = 3/group). G The percentage of released LDH from OGD/R-treated KO or WT-derived neurons was assessed to determine neuronal injury in the presence or absence of HKL (n = 3/group). H Representative scatter plots of apoptotic neurons induced by OGD/R through PI/Annexin V double labeling in the presence or absence of HKL. I Quantification of apoptotic neurons (Annexin V-positive; n = 3/group) in panel G. J Relative activity of MnSOD, CAT, and GSH content in KO or WT-derived neurons after OGD/R treatment with or without HKL (n = 3/group). K Flow cytometry analysis of ROS production in KO or WT-derived neurons after OGD/R treatment in the presence or absence of HKL. L Quantification of ROS production in neurons of indicated groups (n = 3/group). Normalized to the level of WT neurons that were treated with OGD/R. M, N Western blotting was used to determine the effect of HKL on the expression of NMDAR2B, T-AMPK, p-AMPK, PGC-1α and SIRT3 in neurons that were subjected to OGD/R. Relative expression was normalized to the level of the control treatment (n = 3/group). Statistical analysis: mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001
HKL protects mice from SCIRI partially depend on SIRT3After confirming the inhibitory effect of HKL on neuronal apoptosis and oxidative stress in vitro, we then tested the effect of HKL on neurological function recovery in SCIRI mice. HKL treatment (0.2 mg/kg/day, intraperitoneally) [48] was started on the day after surgery and maintained throughout the study. Since HKL is not a SIRT3 specific inhibitor, we also examined the effect of HKL on motor function recovery in KO mice. BMS score, footprint analysis and MEPs results suggested that HKL could effectively improve the recovery of lower limb motor function in SCIRI mice. However, in KO mice, although HKL also partially restored the neurological function of SCIRI mice, this neuroprotective effect was significantly weakened compared with WT mice (Additional file 1: Fig. S4A–E). Further studies suggested that HKL increased the number of Nissl bodies in the spinal anterior horn of SCIRI mice (Additional file 1: Fig. S 4F, G), reduced TUNEL positive cells (Additional file 1: Fig. S4H, I), and improved the reoxidation-reduction imbalance of spinal tissue (Additional file 1: Fig. S4J), but the above effects were also significantly attenuated in KO mice (Additional file 1: Fig. S4F–J). These results suggest that HKL protects mice against SCIRI, and the neuroprotective effect is partly dependent on the expression of SIRT3.
RIPC-HKL combination exhibits better efficacy against SCIRIBased on our findings that HKL is neuroprotective against SCIRI, and that RIPC and HKL protect from SCIRI by independently modulating SIRT3 expression, we assessed if combining HKL and RIPC can enhance their individual protective effects against SCIRI. Therefore, preischemic RIPC and sustained HKL administration after ischemia were combined to treat SCIRI and named combined therapy (Fig. 8A). To test if the effects of two regimens were additive, we compared the efficacy of the combined therapy with that of HKL alone. We first compared the effect of HKL and that of combined therapy on SIRT3 expression in the spinal cord. Western blot results showed that HKL increased SIRT3 level compared with I/R group, and this effect was more obvious when combined with RIPC (Fig. 8B). Similar results were obtained by immunofluorescence (Fig. 8C, D). These results indicated that combining RIPC with HKL was more effective than HKL alone in rescuing SIRT3 expression after SCIRI. Next, we investigated if the upregulation of SIRT3 by HKL alone or the combined therapy restored mouse neurological function. BMS revealed that mice motor function started to improve 24 h after administration with HKL and was further improved by combining HKL with RIPC (Fig. 8E). Moreover, footprint analysis on day 3 after SCIRI revealed that the hindlimb coordination of mice treated with combination therapy were better than that of mice treated with HKL alone (Fig. 8F). The amplitude of MEPs in mice treated with combination therapy was also higher than that treated with HKL alone (Fig. 8G, H). Additionally, Nissl staining revealed that HKL reduced neuronal loss and structural damage (Fig. 8I). TUNEL staining also showed that HKL inhibited I/R-induced apoptosis in mice (Fig. 8J, K). Notably, these neuroprotective effects of HKL were significantly enhanced by combining HKL with RIPC (Fig. 8I–K). Moreover, when compared with HKL alone, the combined therapy further suppressed oxidative stress after SCIRI, as revealed by higher antioxidant enzyme activity and lower MDA levels (Fig. 8L). These results indicate that HKL protects neurons from SCIRI in vivo and that its efficacy is improved by enhanced SIRT3 upregulation when combined with RIPC.
Fig. 8
RIPC combined with HKL achieves better efficacy in mice with SCIRI. A Schematic diagram of experimental design. B Representative immunoblot images showing the effect of HKL and combined therapy on the expression of SIRT3 after spinal cord I/R (n = 5/group). C, D Immunofluorescence was used to detect the effect of HKL and combined therapy on the expression of SIRT3 (in green) in neurons (in red) after spinal cord I/R (n = 5/group). Nuclear was stained with DAPI (in blue). Scale bar = 100 μm. E The mice were functionally scored up to 3d postinjury using the BMS after treatment with HKL or combined therapy (n = 5/group). * p < 0.05, HKL treatment group compared to I/R injury group; # p < 0.05, Combination treatment group compared to HKL treatment group. F Representative footprint images from mice on the third day after injury after treatment with HKL or combined therapy and quantification of the stride length and width (n = 5/group). Blue: frontpaw print; red: hindpaw print. G, H MEP analysis was used as an electrophysiological assessment in mice treated with HKL or combined therapy on day 3 and quantification of the peak-to-peak MEP amplitudes (n = 5/group). I Representative images of Nissl bodies in the anterior horn of the spinal cord and quantification of the number of integrated Nissl bodies per section (n = 5/group). Scale bar = 100 μm. J Representative images of TUNEL-positive apoptotic cells (in red) in spinal cord sections on day 3 after I/R injury and quantification of the number of apoptotic cells in each group (n = 5/group). Neuron was stained with NeuN (in green) and Nuclear was stained with DAPI (in blue). Scale bar = 100 μm. K MnSOD, GSH, CAT, and MDA were measured on day 3 after injury to reflect the level of oxidative stress (n = 5/group). Statistical analysis: mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001
留言 (0)