
記住我


The E3 ubiquitin-protein ligases, the matchmakers in the ubiquitination cascade, are implicated in the regulation of various steps of the autophagic process, the major lysosome-dependent degradation pathway (Yin et al. 2020). Autophagy provides a homeostatic control mechanism and has been found to be defective in cancer (Russell and Guan 2022). Regarding the relevant clinical and therapeutic aspects of autophagy, there is emerging attentions in the exploring the responsible factors affecting the autophagy machinery in the diseases. By a comprehensive databases search, we found that, during recent years, there has been continuously growing evidence that shows a key role of HECT-type E3 ligases, particularly members of Neural precursor cell expressed developmentally downregulated protein 4 (NEDD4) family, in defective autophagy in cancer. Thus, the present review was conducted to address the following questions: (1) which members of the NEDD4 E3 ligase family are implicated in defective autophagy in cancer cells?, (2) which types of cancers are affected?, (3) what is their activity in autophagy in different cancer cells; autophagy inhibitor or autophagy inducer and tumor promoter or tumor suppressor ?, (4) what are their new substrates and molecular mechanisms underlying their effects?, (5) How can they be targeted to conquer different cancers?. To this end, the following sections were arranged. First, an overview of NEDD4 ubiquitin ligases as well as the autophagy process and its role in cancer cells were briefly presented in the following introduction subsections. Afterward, all published data regarding the role of NEDD4 ubiquitin E3 ligases in the autophagy process in cancer have been reviewed and discussed in detail.
NEDD4 ubiquitin E3 ligases: a snapshot view of enzymatic activity and structureE3 ubiquitin ligases are the selective executers in the protein ubiquitination and, thus, implicate in the two major protein degradation pathways, the ubiquitin–proteasome system (UPS) and autophagy (Yin et al. 2020). Ubiquitin is a highly conserved 76 amino acid globular protein. Ubiquitination is a reversible enzymatic conjugation event, which forms an isopeptide bond between the carboxyl group of C-terminal Glyc76 on ubiquitin and the ε-amino group of a Lysine residue on the substrate. Ubiquitin attachment onto target proteins includes a multistep reaction that needs the sequential and coordinated activity of a cascade of three enzymes: a ubiquitin-activating enzyme (E1), a ubiquitin-conjugating enzyme (E2), and a ubiquitin ligase (E3). The E3 ligases transfer activated ubiquitin to a Lysine residue on the target substrate through an interaction involving both the E2 conjugating enzyme and the substrate (Vere et al. 2020).
NEDD4 is a well-known family of the homologous to the E6AP carboxyl terminus (HECT)-type E3 ligases. NEDD4 family comprises nine E3 ligase members: NEDD4-1, NEDD4L, WW domain-containing E3 ubiquitin-protein ligase 1 (WWP1), WWP2, NEDL1 (HECW1), NEDL2 (HECW2), Smad ubiquitin regulatory factors (SMURF)1, SMURF2, and ITCH. NEDD4 members show a highly similar domain architecture consisting of an N-terminal protein kinase C-related membrane/lipid-binding C2 domain mediating attachment of NEDD4 E3 ligases to membrane compartments (Dunn et al. 2004; Plant et al. 2000; Angers et al. 2004; Kumar et al. 1997), two to four tryptophan-tryptophan (WW) domains located in the central part (N-terminus) for the substrate recognition and binding through the interaction with Proline-rich motifs (mainly PPxY) or phosphorylated Serine/Threonine-Proline regions on the target substrate (Kumar et al. 1997; Staub and Rotin 1996), as well as the catalytic HECT domain at the C-terminus (Kumar et al. 1997; Weber et al. 2019; Dodson et al. 2015). The HECT domain directly catalyzes the covalent bond between ubiquitin and target substrates through a two-step reaction: first, they capture the activated ubiquitin from E2 conjugating enzymes in a transthiolation reaction on their catalytic cysteine, and then, the ubiquitin moiety is transferred to a lysine on the substrate. The HECT domain is highly conserved and consists of N- and C-terminal lobes connected by a flexible linker chain. The N-lobe contains the E2-binding site (Fotia et al. 2006), while the C-lobe carries the active-site cysteine catalyzing the thioester bond with the ubiquitin moiety (Verdecia et al. 2003; Huang et al. 1999). The flexible linker permits the C-lobe to move around and assist the ubiquitin transfer from the E2 to the E3 (Weber et al. 2019). In basal status, the NEDD4 E3 ligases can be kept in a catalytically inactive state through an autoinhibitory conformation in which the N-terminal domains (either the C2 or WW domains) interact with the C-terminal HECT domain to luck the HECT activity and prevent substrate or E2 access (Wan et al. 2011; Wiesner et al. 2007; Wang et al. 2019; Zhou et al. 2014).
Proteins can be modified by mono-ubiquitination, as a result of the attachment of a single ubiquitin, or by polyubiquitination through the sequential attachment of ubiquitin moieties on lysine residues. Ubiquitin contains seven internal lysine residues (K6, K11, K27, K29, K33, K48, and K63) that can accept another ubiquitin molecule in subsequent rounds of ubiquitination, finally generating multiple types of polyubiquitin chains (Vere et al. 2020; Xu et al. 2009). Mono- or polyubiquitination and the exact composition of linkage chain determines the distinct fate of the substrates. For example, K63-linked poly-ubiquitylated and mono-ubiquitylated substrates are preferentially degraded by the autophagy/lysosome system, whereas K48-linked ubiquitination is mainly believed to target substrates for proteasome degradation (Kwon and Ciechanover 2017). Of note, K63-linked and K48-linked polyubiquitination compete with each other to activate autophagic proteins in response to stress conditions or to degrade them when the stress situation is resolved, respectively. In particular, K48-polyubiquitination-mediated degradation of autophagy proteins is necessary to terminating the autophagy response (Yin et al. 2020).
An overview of autophagyThe intracellular protein homeostasis is majorly controlled by using two pathways of protein degradation, the UPS and autophagy. Whereas the UPS is the main cellular pathway to degrade short-lived proteins, autophagy is the fundamental catabolic mechanism for degrading and recycling damaged organelles as well as long-lived proteins, protein aggregates, and protein complexes. Autophagy is a conserved self-digestion process, through which cytosolic constituents are sequestered by lipid bilayer vesicles and subsequently transferred to lysosomes for degradation (Cao et al. 2021).
Autophagy exists in a basal (constitutive) as well as stimulated state. Basal autophagy occurs in most cells and tissues under normal physiological conditions to maintain cellular homeostasis. Basal autophagy is also responsible for cellular architectural alterations that happen during development and differentiation (Adelipour et al. 2022; Hu et al. 2019). However, stimulated autophagy can occur in response to cellular stresses such as nutrient or growth factor starvation, high temperature, overcrowding, hypoxia, endoplasmic reticulum (ER) stress, and microbial infection (Cao et al. 2021). In response to such stresses, autophagic degradation is activated to provide biosynthetic demands, reprogram cellular metabolism, and permit cell viability. Notably, starvation induces nonselective autophagy that engulfs any cytosolic constituents. Starvation-induced autophagy permits the cell to recycle nutrients from digested organelles and proteins, thereby maintaining the cellular biosynthetic capacity via providing amino acids for de-novo protein synthesis, and preserving the cellular energy source (ATP) by supplying free fatty acids and amino acids for the Krebs cycle. However, selective autophagic degradation acts by recognition and targeting specific cellular material, such as protein aggregates (aggrephagy), injured organelles (mitophagy for the mitochondria disposal, ERphagy for the ER disposal, and pexophagy for the perioxisomes disposal), as well as intracellular pathogens (xenophagy) (Lamark and Johansen 2021; Janssen et al. 2021).
The autophagy process consists of an orderly set of events: the initiation, phagophore nucleation, sequestration and autophagosome formation, the fusion of the autophagosome with the lysosome, and cargo digestion and recycling. The promotion of autophagy is started with the nucleation of a sequestering membrane, creating a cup-shaped phagophore, which stems from lipid bilayers provided majorly by the ER, but also by endosomes and the Golgi apparatus. A part of cytosol, including organelles, is subsequently engulfed by the elongating phagophore to create a double-membrane vesicle called the autophagosome. Eventually, the outer membrane of the autophagosome merges with the lysosomal membrane to create an autolysosome compartment, where engulfed cytosolic components are degraded by the acidic lysosomal hydrolases (Cao et al. 2021; Sidibe et al. 2022).
The core autophagic machinery relies on autophagy-related (ATG) proteins, which assemble into functional complexes that are recruited to autophagy membrane compartments and work in sequential order to deliver the cytosolic cargo to the lysosomes (Zhou et al. 2022). The master regulator of autophagy is the mammalian target of rapamycin complex 1 (mTORC1), which inhibits autophagy by suppressing the activity of Unc-51-like kinase 1 (ULK1) (Dossou and Basu 2019; Ganley et al. 2009; Hosokawa et al. 2009; Jung et al. 2009; Noda and Fujioka 2015). ULK1 is a serine/threonine kinase and one of the most upstream ATG proteins required for the initiation steps of autophagy in mammalian cells. Under stressful conditions, AMP-activated protein kinase (AMPK) suppresses mTORC1 and activates ULK1 that forms a stable protein kinase complex with autophagic proteins ATG13, ATG101, and FIP20 to initiate autophagy (Dossou and Basu 2019).
The activated ULK complex localizes to discrete sites on the ER and induces the phagophore nucleation via phosphorylating components of the class-III phosphatidylinositol 3-kinase (PI3KC3) complex comprising Beclin-1, Vps34/PI3K, Vps15, ATG14L, UV resistance-associated gene (UVRAG), and Rubicon. Upon phosphorylation, the PI3KC3 complex induces local production of phosphatidylinositol-3-phosphate (PI3P) at ER structures termed omegasomes, where the effector proteins are recruited to initiate the phagophore nucleation (Karanasios et al. 2016).
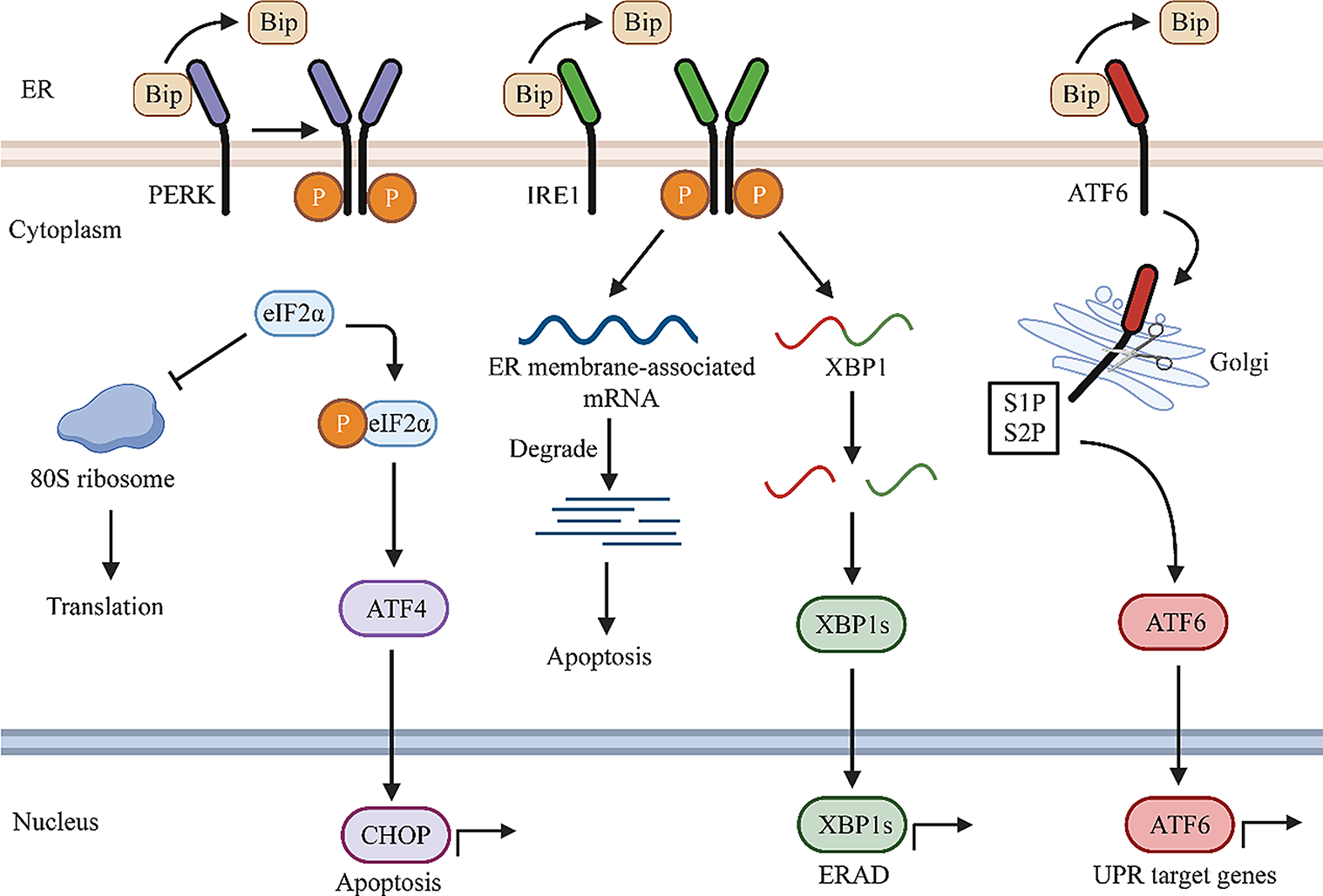
ATG proteins orchestrate the elongation and expansion of the phagophore membrane to the autophagosome. The ATG5 ~ ATG12-ATG16L complex recruits ATG8 [microtubule-associated protein 1 light chain 3 (LC3)] protein that is subsequently lipidated by the sequential activity of ATG4B as well as ATG7 and ATG3 (Carlsson and Simonsen 2015). The lipid conjugation process is initiated via converting LC3 to the active cytosolic isoform LC3-I by the protease activity of ATG4B, in which ATG4B cleaves LC3 to expose the C-terminal Glycine for the subsequent lipidation reaction. ATG3/ATG7 mediate conjugation of membrane-associated phosphatidylethanolamine (PE) and convert LC3-I to lipidated LC3-II (Fig. 1). Indeed, LC3-II is a membrane-anchored form of LC3 that is essential for phagophore elongation and for facilitating the specific recruitment of cargos in selective autophagy (Kabeya et al. 2000; Walker and Ktistakis 2020).
Fig. 1
The schematic view of the role of NEDD4 E3 ubiquitin ligases in the autophagic process in cancer cells
For the selective autophagic degradation, specific autophagy adaptors (receptors), such as p62 [also called sequestosome 1 (SQSTM 1)] (Pankiv et al. 2007; Zheng et al. 2009; Wurzer et al. 2015), autophagy and Beclin 1 Regulator 1 (AMBRA1) (Rita, et al. 2018), Optineurin (OPTN), nuclear dot protein 52 kDa (NDP52) (Heo et al. 2015; Mostowy et al. 2011; Thurston et al. 2009; Muhlinen et al. 2012), and neighbor of BRCA1 gene (NBR1) (Walinda et al. 2014; Riley et al. 2010; Kirkin et al. 2009), attach a ubiquitin-tagged cargo to a nascent autophagosome by concurrently binding the cargo and LC3-II on the sequestering membrane. These adaptors possess a ubiquitin-associated domain (UBA) and an LC3-interacting region (LIR) allowing their binding to ubiquitinated cargoes and LC3-II, respectively (Wurzer et al. 2015; Kirkin et al. 2009; Long et al. 2010; Isogai et al. 2011). Indeed, autophagy adaptors act not only as a bridge between ubiquitinated cargoes and LC3-II by the UBA domain but also as a transporter for cargo delivery to autophagosomes by the LIR domain.
The search strategyA systematic literature search was performed in electronic databases, including Web of Science, PubMed, Scopus, and Google Scholar, without any language restrictions, to find all published articles dealing with the aims of the present study. The search was performed using the terms [(autophagy) AND (E3 ubiquitin ligase) AND (“HECT” OR “homologous to the E6AP carboxyl terminus” OR “NEDD4” OR “Neural precursor cell expressed developmentally downregulated protein 4”) AND (“cancer” OR “tumor”)] in titles and abstracts. In addition, the references of enrolled studies were also manually checked to find other related publications that were potentially missed from database searching.
The role of NEDD4 E3 ubiquitin ligases in the autophagy process in cancerAlthough autophagy can maintain the normal physiological function of cells, excessive autophagy can lead to diseases. The dysregulation of autophagy has been found to exert a role in various human diseases, such as neurodegenerative disorders (Fleming et al. 2022), rheumatic diseases (Celia et al. 2022), muscular diseases, cardiovascular diseases (Gatica et al. 2022), and cancer (Gundamaraju et al. 2022; Ariosa et al. 2021). Autophagy has been known to be a double-edged sword in cancer biology, acting both as a protector of cancer cell survival and a tumor suppressor depending on the cancer type and stage of cancer development. On the one hand, autophagy suppresses tumor growth in the early stages by inhibiting the proliferation of pre-cancerous cells, scavenging toxic molecules correlated with tumorigenesis, and removing damaged organelles. On the other hand, autophagy is able to induce tumor growth and survival in later stages. Tumors are under highly stressful conditions such as hypoxia and nutrient deprivation, and autophagy can increase stress tolerance and provide nutrients to meet the metabolic demands of cancer cells, thereby enhancing cancer-cell survival (Gundamaraju et al. 2022; Ariosa et al. 2021).
In addition, a fine-tuning of autophagic activity is important for the appropriate cellular hemostasis and growth, and defective autophagy with either excessive or low activity can cause cancer cell formation. Accumulating findings indicate that the dysregulation of NEDD4 family E3 ligases can be one of the molecular mechanisms attributed to the dual role of autophagy in cancer cells. A systematic search in different electronic databases indicated that, among HECT-type E3 ligases, members of the NEDD4 family including NEDD4-1, NEDD4L, SMURF-1, SMURF-2, WWP1, WWP2, and ITCH have been investigated in many various cancers with defective autophagy, as reviewed in next sections.
Autophagic-mediated roles of NEDD4-1 in cancer cellsThe ubiquitin E3 ligase NEDD4-1 has been found to involve in the proliferation, migration, invasion, and drug sensitivity of cancer cells. NEDD4-1 exerts the dichotomous roles as an oncoprotein (Eide et al. 2013; Amodio et al. 2010; Wang et al. 2007; Xu et al. 2015; Huang et al. 2017; Li et al. 2015; Sun et al. 2017; Kim et al. 2008a; Jung et al. 2013; Singh et al. 2011; Verma et al. 2017; Yim et al. 2009) and a tumor suppressor (Zhou et al. 2014; Trotman et al. 2007; Liu et al. 2013; Huang et al. 2015; Huang et al. 2020a; Zeng et al. 2014; Platta et al. 2012) in cancer cells (Fig. 2A). The expression of NEDD4-1 has been reported to be elevated in several types of cancers including colorectal (Eide et al. 2013; Kim et al. 2008a), gastric (Kim et al. 2008a), breast (Jung et al. 2013; Singh et al. 2011; Verma et al. 2017; Yim et al. 2009), non-small-cell lung carcinoma (Amodio et al. 2010), bladder, prostate, cervical (Wang et al. 2007; Li et al. 2015), hepatocellular carcinoma (HCC) (Huang et al. 2017), and glioma (Zhang et al. 2013). Its oncogenic or tumor suppressor activities are mainly mediated by ubiquitination of proteins with oncogenic or tumor suppressor functions such as PTEN (Amodio et al. 2010; Wang et al. 2007; Kim et al. 2008a; Jung et al. 2013; Singh et al. 2011; Yim et al. 2009), MDM2 (Xu et al. 2015), CNrasGEF (Zhang et al. 2013; Pham and Rotin 2001), N-Myc and C-Myc (Liu et al. 2013), Her3 (Verma et al. 2017; Huang et al. 2015), SAG (Zhou et al. 2014), AKT (Huang et al. 2020a; Fan et al. 2013), and Ras (Zeng et al.
留言 (0)