
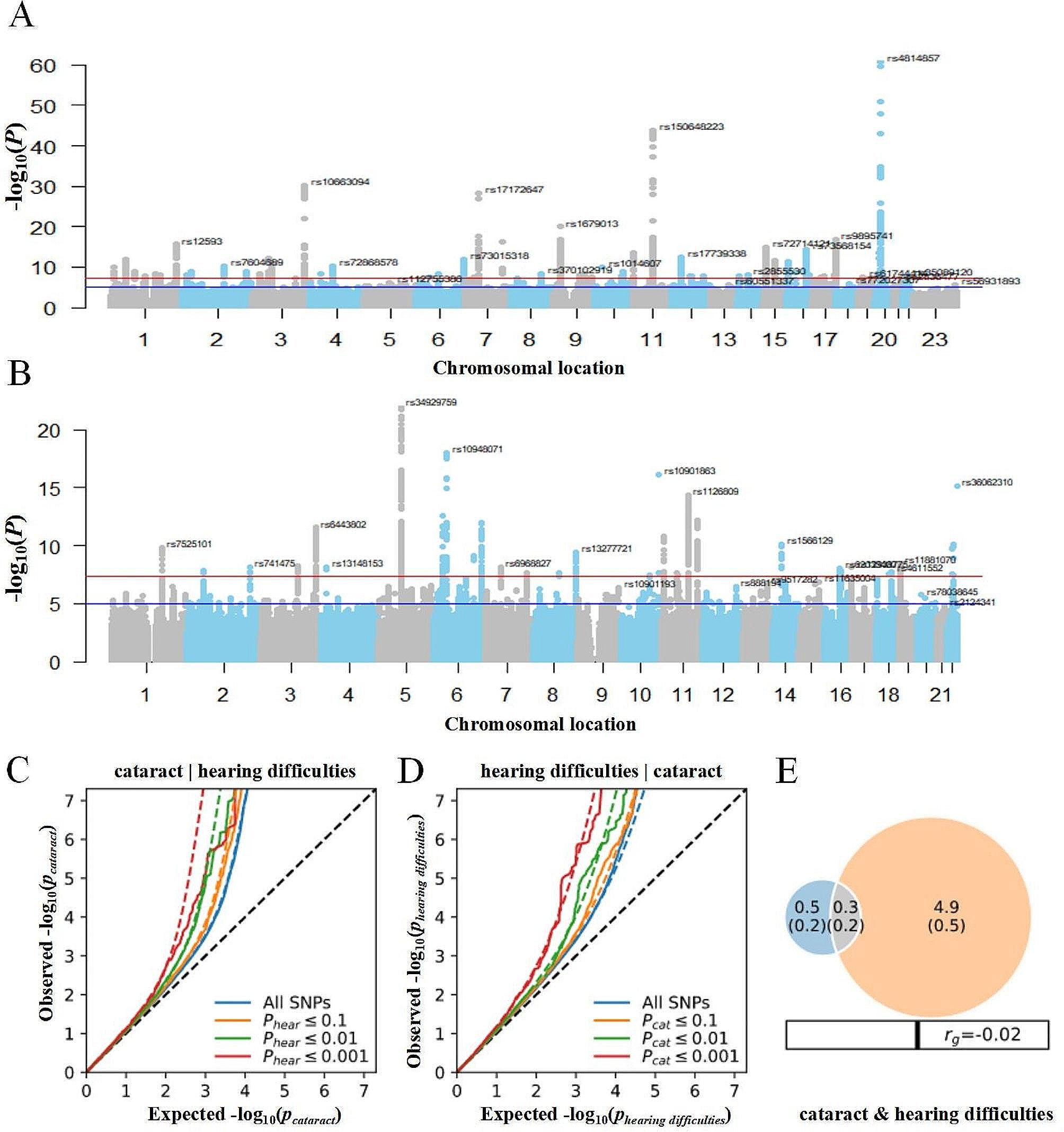
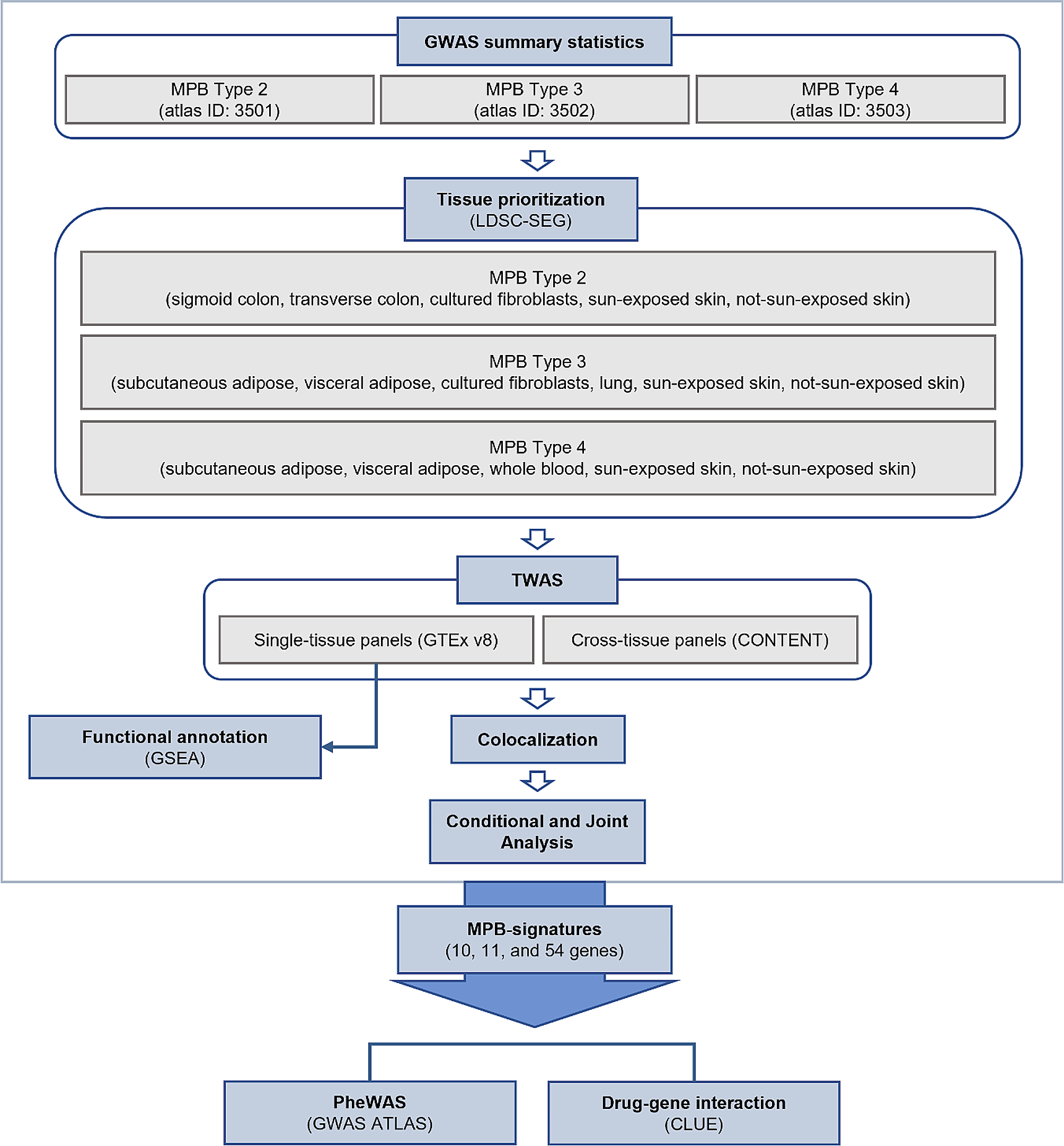
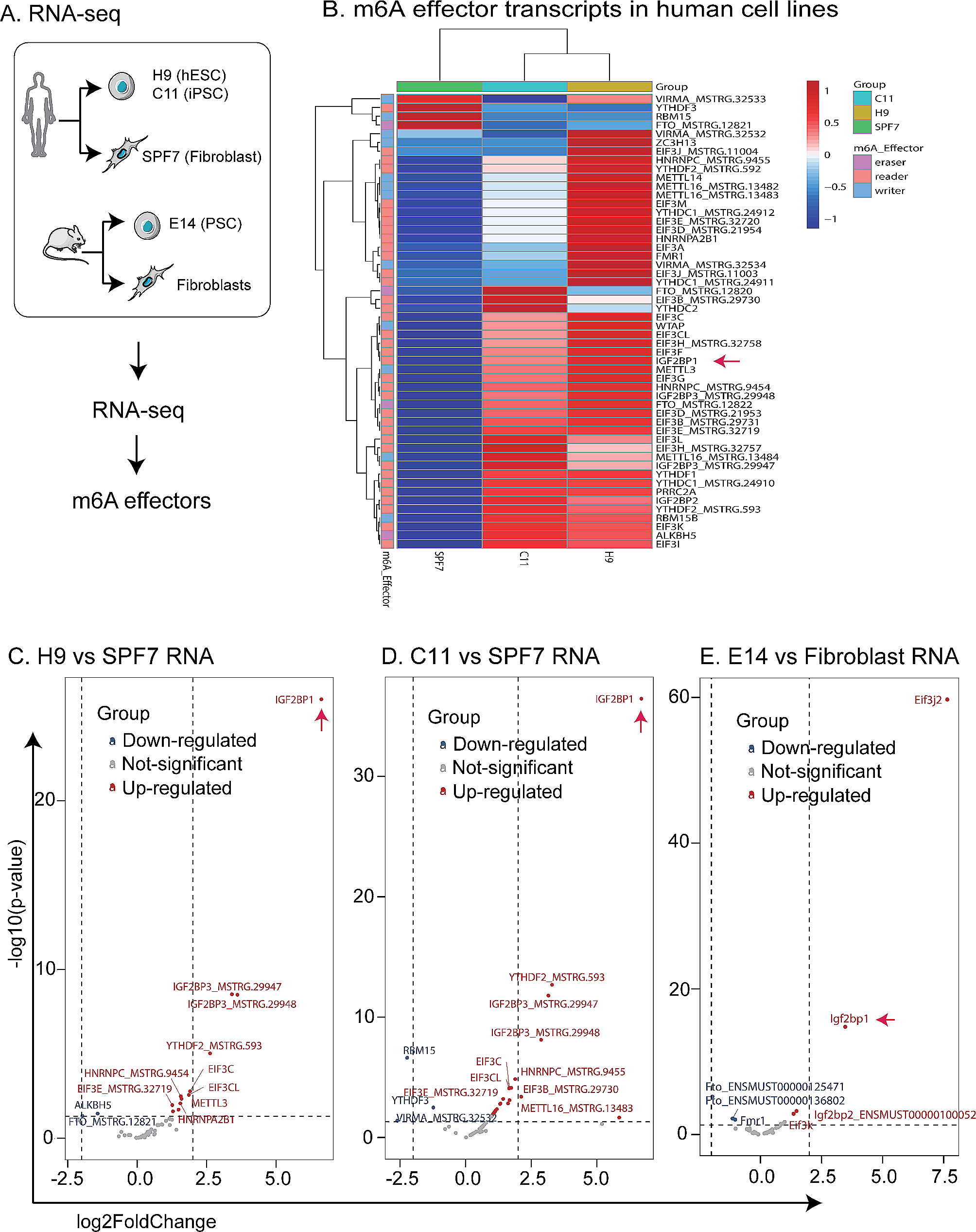
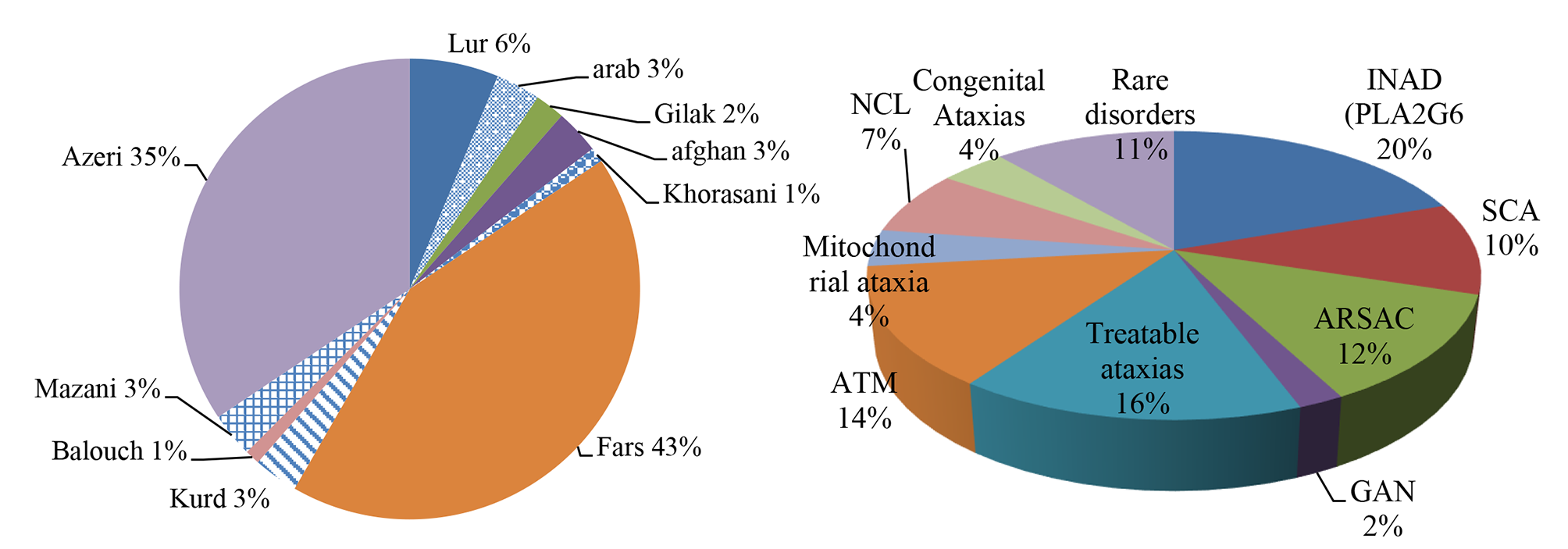
記住我

Chromosomal loci and functional consequences of the 109 genetic risk variants. Autosomal loci of the 60 risk genes associated with severe COVID-19: each dot represents a risk variant, and the dots in the same horizontal line represent the same risk variant but in different genes. The colors of the dots represent risk genes
The distribution of the 109 risk variants after filtration on the human chromosomes is presented in Fig. 2. The ideogram shows chromosomes 1-9, 11, 12, 15, 17, and 19-X contain 109 risk variants with different types of variant function such as intergenic, intronic, and transcript variants that are distributed in 60 genes in the host genome. We found two gene clusters that have an impact on severe outcomes of COVID-19. The first gene cluster contains C4BPA, LOC107985251, CFH, and CD55 located on chromosome 1q31.3-1q.32.2. The loci hold cluster of risk genes are related to immune responses and chemokine and cytokine activity. This cluster contains the regulatory variants rs61821041 and rs61821114, which contribute to downregulation of CD55. Furthermore, rs45574833 is associated with atypical hemolytic uremic syndrome, a condition in which thrombi develop in tiny blood arteries in the kidneys. The regulator of complement activation (RCA) contains many tandemly clustered genes with homologous immune system activities, which are downregulated during COVID-19 infection [15]. In addition, the second cluster of genes: SCN5A, LZTFL1, SLC6A20, FYCO1, CCR9, CXCR6, and XCR1 located on 3p21.31 and 3p22.2 are related to innate immune system activities. For instance, SCN5A in human macrophages functions as a pathogen sensor and modulates antiviral responses and defense [16], the cluster is carried by 50% of South Asian and 16% of European populations, and was previously associated with severe COVID-19 and immune dysfunction inherited from Neanderthals [17, 18]. Therefore, our investigation further emphasizes that this cluster is associated with severe COVID-19.
Moreover, the ideogram of the chromosomal locations of identified risk variants associated with severe COVID-19 illustrates that three haplotypes are located on chromosomes 9, 11, and X. Our investigation, we found that these haplotypes influence the immune response and metabolic system. Loci 9q34.3 holds a haplotype that contains six risk variants located in the ABO gene. This haplotype interacts with FUT1-6 and FUT and negatively influences the biosynthesis of the components of the blood group systems during COVID-19 infection. Additionally, a second IFITM3 haplotype on chromosome 11 was previously associated with higher severity of HIV, Dengue, Ebola, and influenza infections. IFITM3 is an immune effector protein that is essential for both controlling cytokine production and limiting viral replication. The last haplotype shown on the ideogram is located in the TLR7 gene on chromosome X.
In addition, a group of risk variants located within the 21q22.11-21q22.3 loci influence down-regulation of the activity of the IFNAR2 and MX1 genes, which regulate the function of the interferon receptors and B cell-activating factor receptors that are involved in the immune response to viruses.
Furthermore, we found that a group of variants on chromosome 1 (rs1202980, rs60220284, and rs45574833) located in the C4BPA, LOC107985251, and TRIM46 genes are related to breast cancer. Additionally, the rs4341 and rs4343 risk variants located in the AEC gene on chromosome 17 are related to Alzheimer disease and hypertension. Moreover, rs429358 and rs481778 located in APOE and PLEKHA4 genes on chromosome 19 are related to Alzheimer disease. More explanation of disease-variant mapping is provided in the supplementary materials.
At the level of single polymorphisms related to severe COVID-19, most chromosomes hold risk variants related to the blood and immune system that impact the immune response and increase the severity of COVID-19 during infection. For example, the risk variants found on chromosome 2 are related to the innate immune system, which is the first step of defense and interaction. In addition, some risk variants impact the level of biomarkers in blood. For example, the risk variants on chromosome 1 increase blood pressure during infection, so the biomarker (D-dimer) level increases. Furthermore, some of these variants influence metabolic pathways that affect body systems. For instance, the risk variant on chromosome 3 negatively impacts glucose metabolism pathways. Also, the length of the chromosome, gene size, and overlapping might affect variant distribution by chromosome.
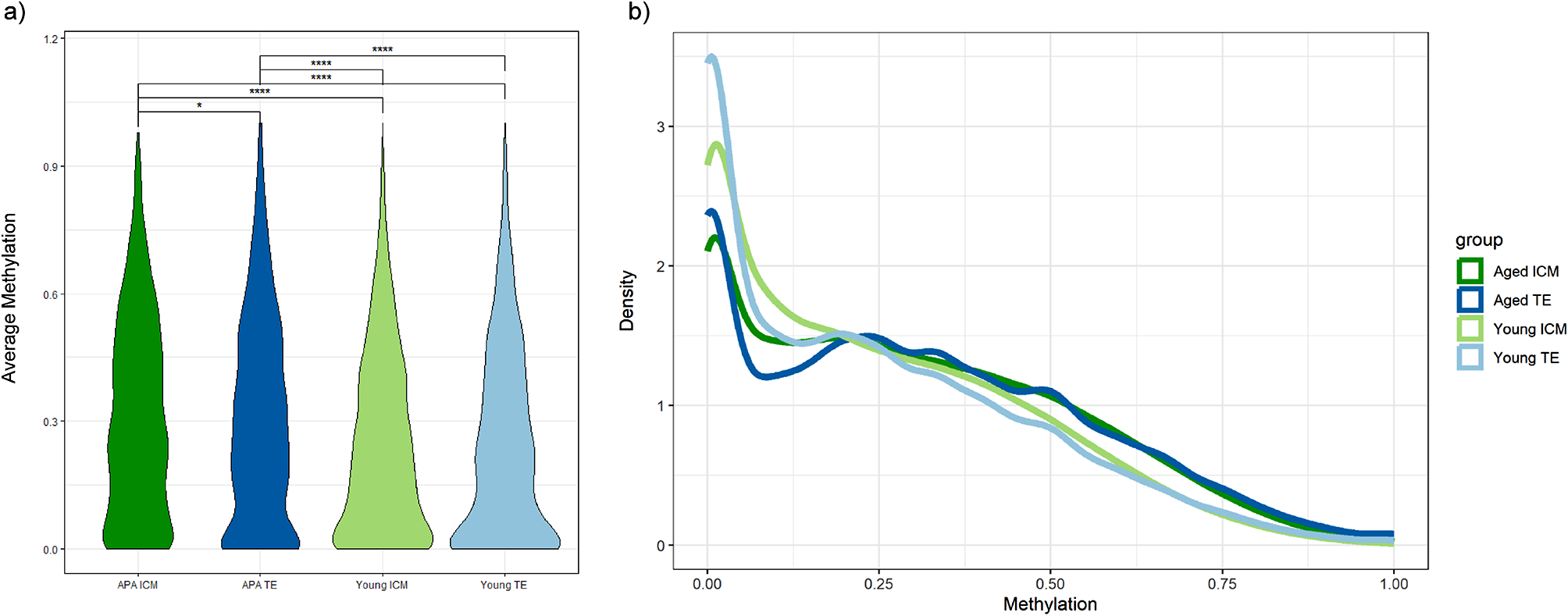
Statistical analysis on the curated dataset of genetic risk variants of severe COVID-19 outcomesIn an initial analysis, our curated dataset of risk genetic variants of severe COVID-19 outcomes was analyzed with the list of risk variants with their reported effects. The data has 109 risk variants with 16 rare variants with minor allele frequencies (MAF) less than 0.01 and 93 common variants, as reported in the reviewed papers. Most of the reviewed papers reported MAF, P value, and other features. However, not all reviewed articles provided the odd ratio (OR) values of variants reported risk variants. Around 50% of the genetic effects of risk variants are not reported in the reviewed articles and need to be calculated. Thus, we re-estimated the additive effect of risk variants by calculating a new OR value for all risk variants using the reported OR and the MAF of each risk variant. Moreover, the association between the MAFs and genetic effects of risk variants of severe COVID-19 is visualized and explained in Additional file 1.
Fig. 3
The additive effects of common risk variants on severe COVID-19 outcome per genes. The scatter plot shows the additive effects of common risk variants on severe COVID-19 outcomes per gene. Each point corresponds to the additive effect of the risk gene that has been calculated based on cumulative values of reported ORs and MAF. Each gene hosts at least one reported risk variant
The associations between risk variants and their effects in developing severe outcomes of COVID-19 per gene were established using an additive genetic model estimating the OR of severe COVID-19. The cumulative values of reported OR and MAF of the accumulated risk alleles in a particular gene were calculated and used as a combined effect to estimate the additive effects per gene. Figure 3 shows a scatter plot of the additive effects of risk variants on severe COVID-19 outcomes per gene. Moreover, the statistical method for estimating the genetic effects of severe COVID-19 is explained in Additional file 1.
Enrichment analysis of genetic risk factors for severe COVID-19Our curated dataset represents variant profiles that provide descriptive information on risk variants. In our analysis, we named each variant associated with severe COVID-19 in the list as a risk variant and its host gene, as a risk gene causally associated with increased mortality in COVID-19. Furthermore, we named the proteins encoded by risk genes risk proteins. The list of genetic risk variants associated with severe COVID-19 is displayed in Additional file 2: Table S1. Below, we present the enrichment analysis of the genetic risk factors associated with the severity of COVID-19 at different levels: variants, genes, and proteins.
Variant level: COVID-19 risk variants and disease associationWe found 46 risk variants in coding regions and 63 located in non-coding regions. In non-coding regions, three variants are located in the intergenic region between genes. Fifty-two of the 63 non-coding region variants are intronic variants, and the remainder are transcript variants occurring within intron regions [19, 20]. The distribution of the functional consequences of 109 filtered risk variants in human DNA is shown in Fig. 4.
Fig. 4
Distribution of functional consequences of the 109 genetic risk variants in the human genome. The height of each bar represents the total number of risk variants in genetic regions, and the light gray color illustrates the number of risk variants associated with other diseases
Table 1 List of the risk variants for severe COVID-19 and their associated diseases and biomarkersTable 1 displays the variant-disease associations for 41 risk variants. Seven risk variants are related to metabolic disorders such as diabetes mellitus, hyperglycemia, and blood protein levels. Three of the intron variants have an impact on ABO protein synthesis, which influences the red cell count and is associated with metabolic diseases such as diabetes and venous thromboembolism (VTE) [21, 22]. The rs12683493 risk variant changes glycosylation and causes von Willebrand disease [22, 23].
Moreover, four risk variants are linked with cardiovascular disorders such as cardiac arrhythmia, long QT syndrome, and triple vessel diseases. Three risk variants are associated with immune dysfunction; for example, white blood cell disorders, complete blood count disorders, and rheumatoid arthritis. Three risk variants are mapped to gastrointestinal diseases, such as Crohn’s disease and inflammatory bowel disease. Four risk variants are associated with cancer such as prostate cancer and Kaposi’s sarcoma. Another five risk variants are related to severe symptoms in infectious diseases namely, tuberculosis and severe influenza. One variant is related to respiratory disorders and mapped to severe asthma.
Gene level: COVID-19 risk genes and disease association Fig. 5
Risk gene set enrichment analysis of the 60 risk genes related to severe COVID-19. A The top ten ranked gene expression scores in human normal tissues and systems: the vertical axes represent the top ten systems based on ranked scores. Each bar represents a system, and the slots inside the bar represent the percentage of the risk genes expressed in various human tissues or cells based on the ranked gene expression scores. The horizontal axes represent the ranked score from 0 to 5. B The top ten human compartments and tissues with the highest numbers of enriched risk genes based on gene ontology analysis
The results of gene set analysis of the COVID-19 risk genes are shown in Fig. 5. The gene expression of the top ten ranked systems in normal tissues and cells derived from our enrichment analysis of the risk gene set is shown in the bar chart in Fig. 5A. Most of these risk genes are highly expressed in blood cells in the hematopoietic system, with a ranked score of 3.5 related to the immune response and viral interaction. The second system is the musculoskeletal system with a ranked score of 3, with some of the risk variants involved in blood circulation. The other three related systems are the renal, reproductive, and neuro systems and are approximately similar with ranked scores of 2.5. The risk genes for the respiratory system and the lungs only have a ranked score of 1.5.
Screening the list of risk genes can help to comprehend the organs and systems affected in severe COVID-19. However, the presence of gene expression does not necessarily mean that the gene is connected functionally to a network related to its expressed system or tissue. For example, three genes out of 60 risk genes are RNA-encoding genes affiliated with the ncRNA class LOC105378861, LOC107986083, and LOC107985251. The phenotype of LOC105378861 is related to the levels of tissue factor activity in blood and increased D-dimer levels in patients with COVID-19 [24]. However, no information is available about LOC107986083 and LOC107985251.
Figure 5B illustrates the number of risk genes that are enriched in human compartments and tissues. Mainly, most risk genes are enriched in the immune system and are located on the cell surface and related to the receptor of type I interferon or viral assembly compartments.
Fig. 6
Gene set and disease analysis of the 60 risk genes related to the severity of COVID-19 outcomes. The top diseases were mapped to the risk genes based on the OMIM and Alliance-DISEASES databases. The circle size represents the number of genes associated with the disease, and the range of colors (high-significant level: red, low-significant level: green) represents the FDR scores for the disease associations
The results of the risk gene analysis with regard to disease associations are illustrated in Fig. 6. The top most-enriched risk genes mapped to disease based on the Mendelian Inheritance in Man (OMIM) database [25]. Inherited Alzheimer disease was the top disease that mapped to the risk genes, followed by long QT syndrome, myocardial infections, metabolic diseases, and lung dysfunction.
Protein level: COVID-19 risk proteins and molecular functional analysisWe found 56 proteins involved in the development of severe COVID-19. We applied GO enrichment analysis of the risk proteins derived from our dataset after eliminating three genes that did not encode proteins. This analysis showed 24 proteins are involved in the immune system.
Fig. 7
GO enrichment analyses of the 56 risk proteins related to severe COVID-19 outcomes. A The top ten significant biological processes and hierarchical correlation clustering trees of the biological processes enriched with the risk proteins. B The top ten significant molecular functions and hierarchical correlation clustering trees of the molecular functions enriched with the risk proteins. C The top ten significant cellular components and hierarchical correlation clustering trees of the cellular components enriched with the risk proteins
Figure 7A shows the risk proteins are mainly enriched in the following biological processes: negative regulation of complement activation, response to interferon-beta, type I interferon signaling pathways, and cellular response to type I interferon. In terms of molecular function, Fig. 7B illustrates the risk proteins are highly enriched in peptidyl-dipeptidase activity, and chemokine binding with high false discovery rate (FDR) scores; these activities are involved in the immune system response. In addition, the results of the enrichment analysis of risk proteins in terms of cellular components are shown in Fig. 7C. The risk proteins are enriched in blood microparticles. Furthermore, the clustering trees summarize the correlation between the significant molecular pathways and functions. The pathways with many shared risk proteins cluster together in one branch. More significant P values are indicated by larger circles.
Table 2 Main host biological processes associated with the 56 risk proteins that contribute to the development of severe COVID-19Table 2 displays the groups of risk proteins involved in biological process pathways related to immune and metabolic activities that contribute to the development of severe COVID-19 symptoms.
Summary of the enrichment analysis of risk factors for severe COVID-19In summary, after applying a comprehensive enrichment analysis of our curated dataset of 109 risk variants associated with severe COVID-19 at different levels (variants, genes, and proteins), we mapped the genetic factors to related diseases to infer the relationships between severe COVID-19 and future complications. Based on the chromosomal distribution and risk variant-disease mapping, we identified three clusters of genes related to immune, hematopoietic, and metabolic dysfunction. Furthermore, three haplotypes contribute to hematopoietic and immune system complications. We found a haplotype of contiguous variants that contribute to regulatory functions or are associated with other diseases that cause severe COVID-19 symptoms. Six risk variants are located within contiguous loci in ABO involved in glycosylation metabolism. In addition, it is notable that the TMRSS2 and ACE2 receptors contain a massive number of contiguous variants that may increase the susceptibility of host cells to viral infection. Numerous groups of variants on chromosomes 21 and X influence the antigenic response of SARS-CoV-2 variants. Moreover, polymorphisms have an influence on host immune recognition and the susceptibility and intensity of the immune response to the SARS-CoV-2 virus [7, 24, 26,27,28,29]. Hence, we applied enrichment analysis on the set of host risk variants to evaluate gene differentiation and discover biomarkers for tissues and cells from the set. The results show that most genes are expressed in both the hematopoietic and immune systems. Furthermore, the molecular functions and biological processes of the list of proteins encoded by risk genes illustrate these proteins are involved in immune responses and metabolic activities.
Molecular network construction and integrationWe eliminated four genes that did not encode proteins. Then, we constructed protein–protein interaction networks for 56 risk proteins that contain a total of 939 interacting proteins, including 48 risk proteins with 939 interactions, and seven orphan proteins that did not interact with any protein. We integrated all constructed networks for the risk proteins and obtained 24 connected PPI networks and seven orphan proteins. In addition, Table 3 displays the main systems and host tissues involved in the constructed networks and affected by the risk factors for severe COVID-19. More details of the PPI networks and related functions are displayed in Additional file 3.
Table 3 Summary of the 24 molecular networks constructed using the genetic risk factors for severe COVID-19Based on the functional analysis above, most networks are related to the blood and immune systems. However, there is no apparent interaction that connects the proteins in these networks, which points to a missing interaction or link. Thus, to highlight such missing links, we adopted another approach to correlate the constructed networks with common diseases that are related to the risk variants hosted in these networks.
Fig. 8
Molecular networks of the 56 risk proteins mapped with the 109 risk variant-disease associations. The red nodes represent COVID-19 risk proteins. The gray nodes represent human proteins that interact with risk proteins. A Twenty-four connected networks. Each connected network has at least one interaction with another protein. Networks 1 to 3 contain more than one risk protein, and Networks 4 to 22 are isolated networks has one risk protein. B Seven orphan risk proteins did not have any protein interaction with other human proteins. The dashed lines link risk variants with linked networks. The different colors of squares illustrate the disease mapping based on the risk variant-disease mapping and the similarity of the molecular functions between the constructed networks
Figure 8 illustrates the 24 PPI constructed based on the results of the risk variant-disease mapping in order to indirectly infer the common molecular functions between the constructed PPI networks. The variant-disease mapping is listed in Additional file 2: Table S2.
According to our investigation of the 24 constructed PPI networks, we infer that the molecular functions of the risk protein interactions are involved in three main host systems: the immune, metabolic, and cardiovascular systems.
In terms of risk variant mapping, Networks 2, 7, and 21 have a common risk variant rs13168774 located on chromosome 5 and associated with respiratory inflammation and metabolic disorders. Moreover, Networks 18, 19, 23, and 24 have a common risk variant rs11385942. These networks have common molecular functions related to immune responses. With reference to the similarity of biological processes and molecular functions between networks, Networks 1, 2, 9, 10, 15, 16, and 19-24 are related to the immune and hematopoietic systems. Networks 3, 7, 13, 14, and 17 are related to the metabolic system. Networks 4, 5, 17, and 18 are related to the cardiovascular system. Networks 3 and 17 are related to the urinary system. Network 11 is related to the nervous system. Network 12 is related to the endomembrane system.
Based on the evidence gleaned from the molecular function analysis of the constructed networks and risk variant-disease mapping, we inferred the hidden PPIs between unconnected networks. The molecular function analysis revealed several hidden pathways. For example, we found unconnected Networks 1-3, 6-10, and 12-24 have similar molecular functions and biological processes related to the immune system. In addition, we inferred unconnected Networks 1-5, 13, 14, and 17-16 share common pathways related to the cardiovascular system and diseases. Moreover, Networks 1, 3, and 11 have common functions and diseases related to the neuron system. Furthermore, based on the disease mapping similarity, we derived that unconnected Networks 1-3, 15-17, and 21 have pathways related to the renal system. Thus, we infer hidden interactions could connect these networks.
Molecular pathways of constructed networksOut of the 24 constructed networks, we analyzed the pathways in Network 1, the largest network that has the highest number of risk proteins (n = 16) and pathways related to the host immune system and contains 511 interactions. Based on the top ten functional pathways of Network 1, we found that eight of the ten molecular pathways in Network 1 are related to cytokine signaling and responses in the immune system and SARS-CoV-2 responses in the innate immune system. The remaining three pathways are related to Influenza A, measles, and Hepatitis C infections. Figure 9 shows the molecular pathways for the risk factors from Network 1; this network is related to the immune system, which confirms that the majority of pathways in Network 1 are related to immune responses. Moreover, the functional pathways in Network 2 contain five risk proteins and 75 interactions that are mainly related to the innate immune system. We found that Network 2 has eight significant pathways with P values higher than 2.6e−13 related to the complement and coagulation cascade pathways. The complement system is a central component of the innate immune system.
Fig. 9
Molecular pathways between the 16 genetic risk proteins of severe COVID-19 in Network 1. The top ten significant molecular pathways between the genetic risk proteins and other host proteins are mainly connected to the host immune system. Network 1 is the largest network and contains the highest number of risk proteins compared to other networks
However, some constructed networks are not related to the immune system. For instance, Network 3 contains three risk proteins and 27 interactions and is related to the metabolic and renin-angiotensin systems, which play an essential role in the regulatory functions and processes of renal, cardiac, and vascular metabolism and physiology. We found that Network 3 has five significant pathways with P values < 5e−08 related to peptide hormone metabolism and protein digestion and absorption, which are involved in metabolic processes and systems. In addition, Network 4 contains three risk proteins and 27 interactions related to the cardiovascular system. We found that all top ten pathways in Network 4 are involved NOTCH signaling and the intracellular domain of NOTCH regulates transcription of genes related to cardiac development. Figure 10 shows the molecular pathways of the remaining constructed Networks 2-24. More details of the molecular pathways between the proteins in these networks and pathway analysis of the remaining constructed networks are demonstrated in Additional files 3 and 4.
Fig. 10
Overview of the molecular pathways of the risk proteins in the constructed networks related to the host metabolic, cardiovascular, and other systems. The figure demonstrates the significant pathways related to severe COVID-19 outcomes. More details of the remaining constructed networks are provided in Additional file 4
Host genetic risk factors and the SARS-CoV-2 pathwayAfter applying molecular function enrichment analysis and disease mapping to risk factors related to severe COVID-19, we found that the majority of pathways are related to the immune system, which suggests that the risk factors associated with COVID-19 are mostly present in proteins involved in the immune system. A minority of networks that have three or more risk proteins, such as Networks 3 and 4, are related to the metabolic and cardiovascular systems. This evidence indicates that the genetic risk factors associated with severe COVID-19 are involved in different host systems that cause multi-organ dysfunctions.
Fig. 11
Host-SARS-CoV-2 pathogen interaction pathway in COVID-19. The location of the 56 genetic risk proteins in the main molecular pathway involved in the host response to SARS-CoV-2 derived from KEEG database. The proteins involved in this pathway are components of different constructed molecular networks, such as Network 1, 3, and 20
Figure 11 shows the location of the risk proteins in the main host-virus pathway, which contains ten risk proteins from different constructed molecular networks. For instance, IL-6, TLR7, OAS, and IFNAR from Network 1 and TMPRSS2 from Network 20 are mainly related to immune system and cytokine and interferon signaling. In addition, ACE and ACE2 from Network 3 are related to the metabolic system and processes.
留言 (0)