
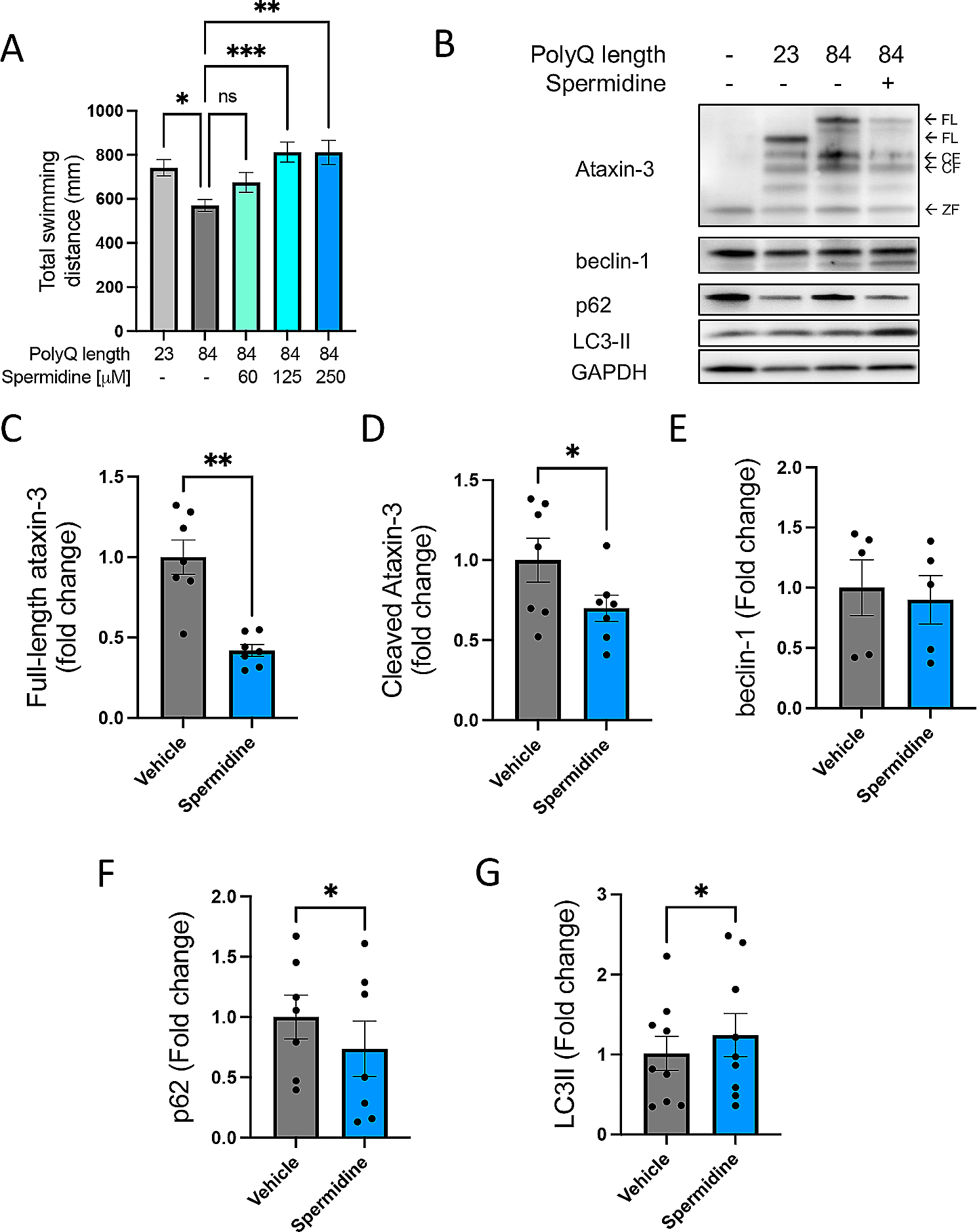
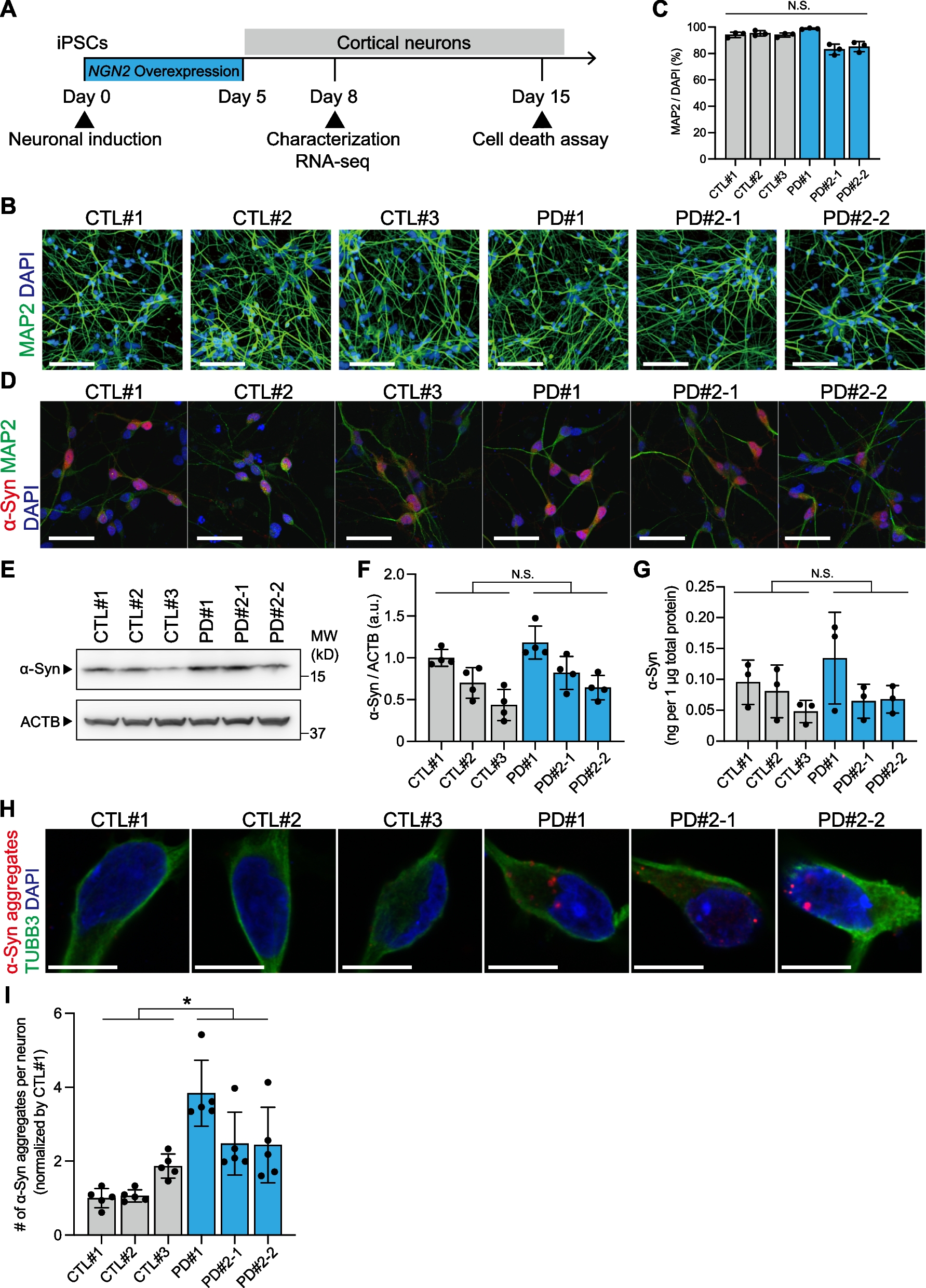
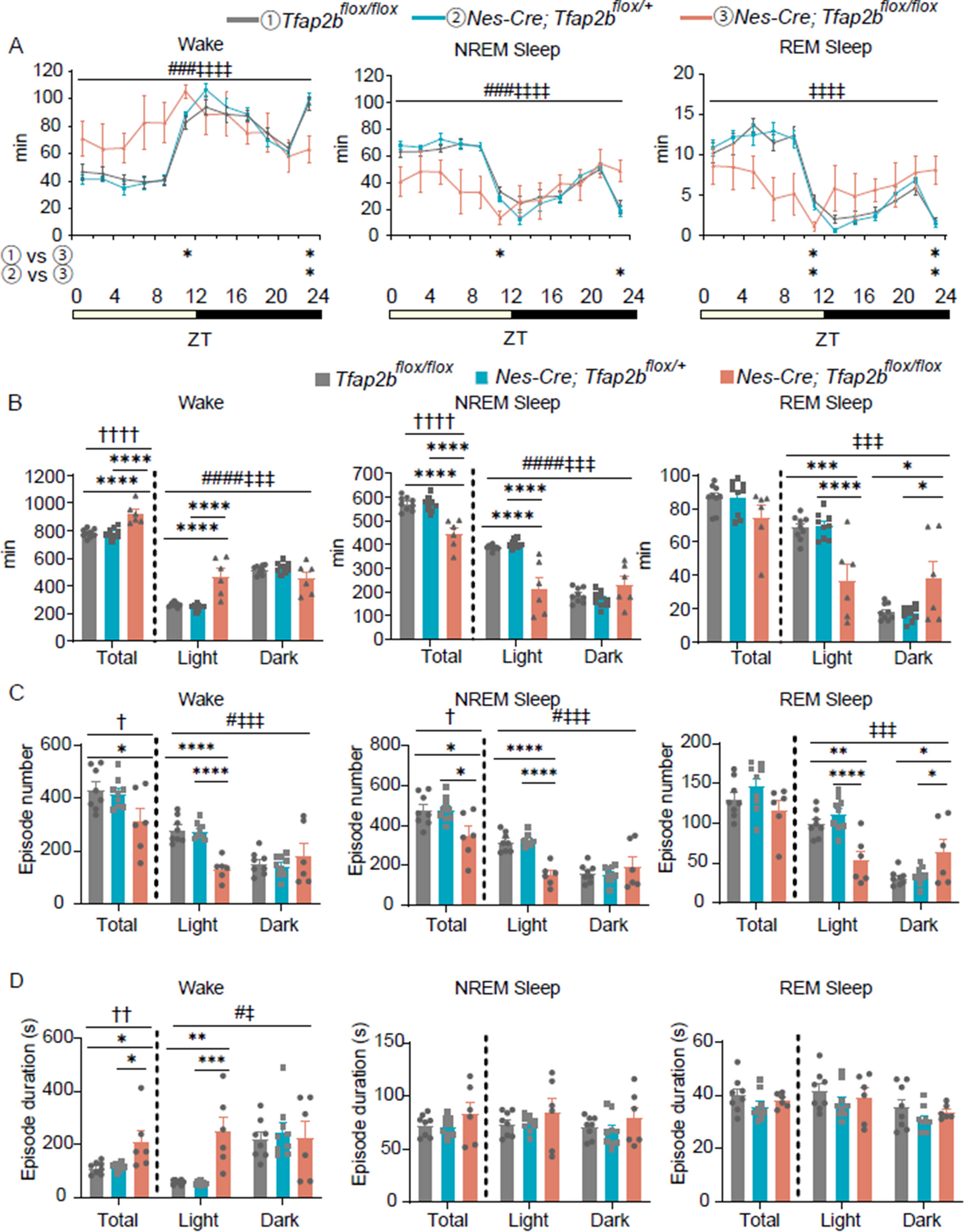
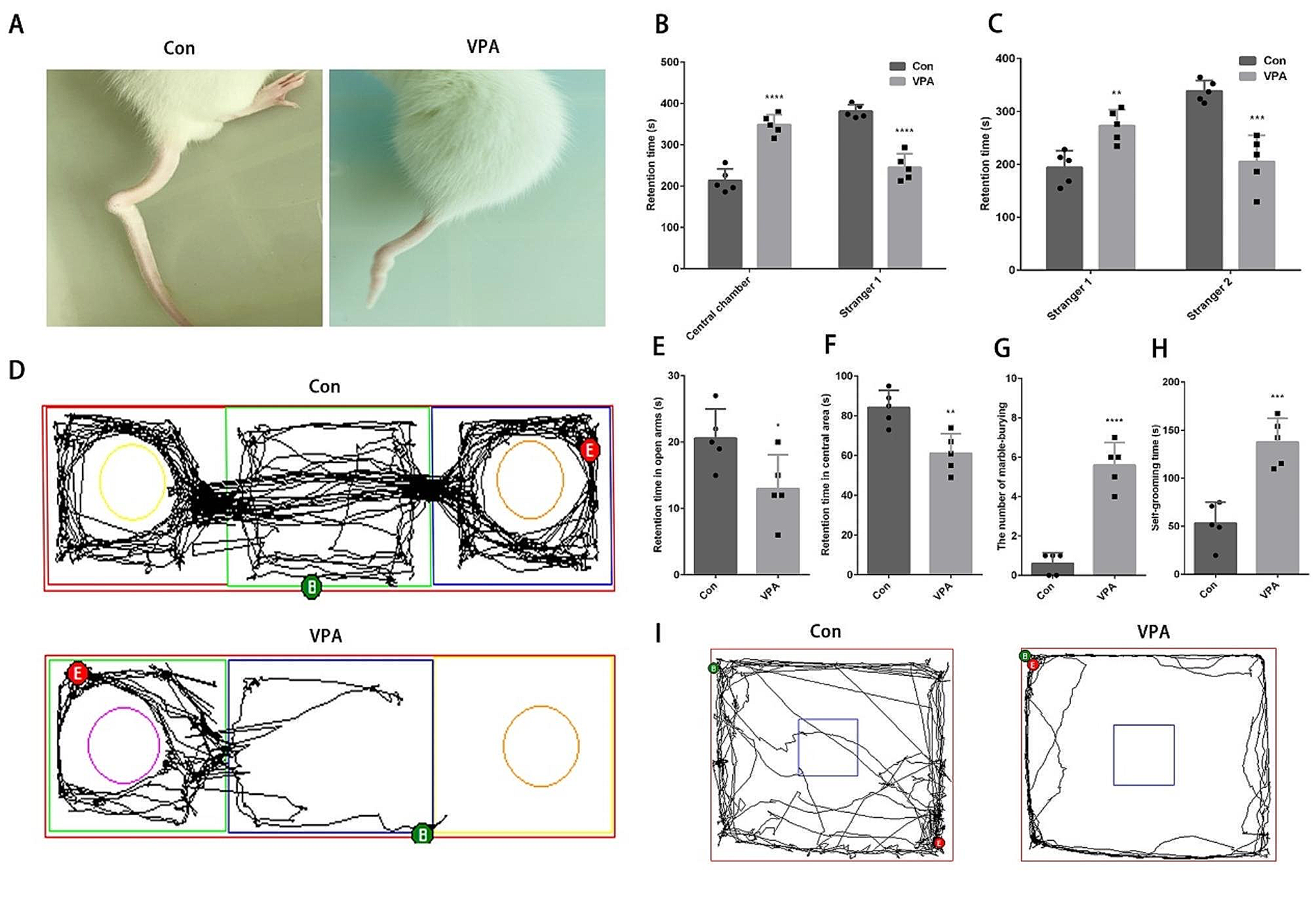
記住我




The participants, all from Guangdong Province, had similar dietary habits and were given standard dietary guidance for three days before the study. There were no significant differences in age and gender between the SCI group and the control group, which minimized the influence of confounding factors on the study results. Detailed data for the SCI patients are shown in Table 1.
Table 1 Comparison of baseline data between patients with SCI and healthy controlsThe gut microbiota profiles of the two groupsTo investigate whether the gut microbiota profile was changed in patients with SCI, 16S rRNA gene sequencing was performed on fecal samples from both the SCI and Control groups. A total of 1,900,745 sequences were obtained. The OTU similarity level for index assessment was 97%. The richness and evenness of the gut microbiome of the two groups were analyzed using rank–abundance curves (Fig. 1a). The rarefaction curve had obvious asymptotes, the OUT coverage was 98.98% (Fig. 1b), and the core species curve had leveled off (Fig. 1c). These results indicated that the community was adequately sampled. Beta diversity analysis (PCoA and PLS-DA) results showed a significant separation of the gut microbiota between the SCI and Control groups (Fig. 1d, e). ANOSIM analysis demonstrated that the gut microbiota composition of the two groups was statistically different, suggesting that SCI induced gut dysbiosis (Fig. 1f).
Fig. 1
Detection of fecal sample quality and differences in gut microbiota composition between groups. a Rank–abundance curves for fecal samples from the control and spinal cord injury (SCI) groups. The abscissa represents the rank of the number of operational taxonomic units (OTUs) and the ordinate represents the relative percentage of OTU number. b Sobs index of rarefaction curves at the OTU level between the two groups of samples detected using a 97% similarity threshold. c Core curves. The horizontal axis represents the number of observed samples and the vertical axis represents the number of all core species at the OTU level. d Principal coordinate analysis (PCoA) score plots. e Partial least squares discriminant analysis (PLS-DA) score plots. f Weighted UniFrac distances
Further analysis was performed at different taxonomic levels based on the annotated species results. Firmicutes, Actionbacteriota, Bacteroidetes, and Proteobacteria were the most abundant phyla among the gut microbiota of both groups (Fig. 2a). In addition, compared with the healthy controls, the abundance of Synergistota was significantly increased in patients with SCI, whereas that of Firmicutes was significantly decreased (Fig. 2b). At the genus level, the abundance of UBA1819 (LDA = 4.54) and Eggerthella (LDA = 3.88) was significantly increased in SCI patients relative to that in the healthy controls. The results also showed marked decreases in the abundances of Blautia (LDA = 4.51), Faecalibacterium (LDA = 4.57), Escherichia–Shigella (LDA = 4.41), Agathobacter (LDA = 4.13), Collinsella (LDA = 4.04), Dorea (LDA = 3.88), Roseburia (LDA = 3.97), Lachnospiraceae_ NK4A136 group (LDA = 3.82), Fusicatenibacter (LDA = 3.80), Holdemanella (LDA = 3.87), Ruminococcus (LDA = 3.81), UCG-002 (LDA = 3.74), and Clostridia_UCG-014 (LDA = 3.84) (Fig. 2c).
Fig. 2
Alterations in the gut microbiota at the phylum and genus levels between the two groups. a The six most abundant species at the phylum level in the two groups. b, c Microbiota displaying significantly different abundances at the phylum and genus levels (*p < 0.05, **p < 0.01, ***p < 0.001). d A cladogram of linear discriminant analysis (LDA) effect size (LEfSe) results in the Control and spinal cord injury (SCI) groups. e Histogram of the LDA scores calculated for a differential abundance of functional profiles in the two groups. A LDA score cutoff of 3.0 was used to indicate a significant difference. Different colors represent different groups. f Correlation between fecal microbiota structure and samples
To further determine the specific gut microbiota components associated with SCI, LEfSe analysis was used to identify the gut microbiota components of both groups. The results revealed 68 components with different classification levels, 20 of which were enriched in SCI patients and 48 in the Control group (LDA > 3; p < 0.05, Fig. 2d, e). Classification results showed that the 20 species enriched in the SCI group belonged to the phyla Firmicutes (n = 16), Proteobacteria (n = 2), Actinobacteriota (n = 1), and Bacteroidota (n = 1), while the 48 species enriched in the Control group belonged to the phyla Firmicutes (n = 36), Proteobacteria (n = 6), Actinobacteriota (n = 4), and Bacteroidota (n = 2). The correlation between fecal microbiota structure and fecal samples is shown in Fig. 2f.
The serum metabolite profile of both groupsTo determine the extent of metabolic disorder resulting from SCI, untargeted metabolomics analysis was used to evaluate the differences in metabolite abundance between serum samples of the SCI group (n = 10) and those of the Control group (n = 10). In the metabolic profiles of all the samples, 5,039 positive and 4,894 negative model features were identified. As shown in Fig. 3a, when the relative standard deviation (RSD) was < 0.3, the peak proportion was > 70%, indicating that the sample size was appropriate. A comprehensive multivariate statistical analysis of cations and anions was undertaken using PLS-DA and OPLS-DA. In the PLS-DA (Fig. 3b, c) and OPLS-DA (Fig. 3e, f) score plots, a significant separation was observed between the Control and SCI groups, indicating that SCI led to metabolic dysfunction. Furthermore, permutation tests showed that the PLS-DA (Fig. 3d) and OPLS-DA (Fig. 3g) patterns had good reliability.
Fig. 3
Changes in serum metabolite abundance in the Control and spinal cord injury (SCI) groups. a Relative standard deviation (RSD) distribution plot. b, c Partial least squares discriminant analysis (PLS-DA) score plots in positive ion mode and negative ion mode, respectively. d Model verification map of PLS-DA (permutation test). e, f Orthogonal PLS-DA (OPLS-DA) score plots in positive ion mode and negative ion mode, respectively. g Model verification map of OPLS-DA (permutation test)
A total of 1511 differential metabolites (p < 0.05, VIP score > 1) were detected. Furthermore, 41 named differential metabolites were quantified. Forty-one metabolites exhibited significant differential abundance between the SCI patients and healthy controls, 18 of which were upregulated and 23 downregulated (Fig. 4a). The differentially abundant metabolites are listed in Table 2. The metabolites exhibiting significant differential abundance between the two groups are shown in the cluster heatmap in Fig. 4b.
Fig. 4
Differential metabolite extraction and KEGG pathway enrichment analysis. a Volcano plot of the differentially abundant metabolites. The abscissa is the multiple change value of the expression difference of metabolites between the two groups, and the ordinate is the statistical test value of the expression difference of metabolites (p-value). Each point in the figure represents a specific metabolite. b Heatmap of the differentially abundant metabolites between the two groups (variable importance in projection [VIP] scores > 3, p < 0.05). The color represents the relative abundance of the metabolites in the samples. c Level 1 and 2 KEGG pathways related to the differentially expressed metabolites. The ordinate is the name of the level 2 pathway and the abscissa is the number of metabolites related to that pathway. Different colors represent different level 1 pathways. d KEGG pathway enrichment column chart. The abscissa is the name of the level 3 pathway. CP cellular processes, EIP environmental information processing, GIP genetic information processing, HD human diseases, M metabolism, OS organismal systems
Table 2 Differentially expressed serum metabolites between patients with SCI and healthy controlsDifferential metabolites and KEGG pathway enrichment analysisWe next applied KEGG pathway enrichment analysis to the 41 named differential metabolites. The results suggested that the altered metabolites were mainly related to amino acid metabolism, digestive system, nucleotide metabolism, and membrane transport (Fig. 4c). The 41 metabolites were enriched in 20 KEGG pathways (p < 0.05). Histidine metabolism: M and FoxO signaling pathway: EIP were the two most significantly enriched pathways (p < 0.01, Fig. 4d).
Analysis of the correlations among gut dysbiosis, altered serum metabolites, and clinical parametersSpearman’s correlation was used to investigate the relationship between gut microbiota and metabolites. The relationship between the 20 most differentially expressed metabolites and the 20 most differentially abundant gut microbiota at the genus level was analyzed in patients with SCI (Fig. 5a). Significant correlations were found between UBA1819 and uridine (C = 0.609, p = 0.004), Lachnospiraceae and hypoxanthine (C = 0.595, p = 0.006), Blautia and PC (18:2/0:0) (C = 0.659, p = 0.002), and Akkermansia and kojic acid (C = 0.628, p = 0.003). Meanwhile, the abundance of Akkermansia was significantly and negatively correlated with that of (5--2-hydroxyphenyl) oxidanesulfonic acid in serum (C = 0.609, p = 0.004).
Fig. 5
Analysis of the correlation among the gut microbiota, serum metabolites, and clinical parameters. a Heatmap of the correlations between differentially abundant species and metabolites (associations between 41 differentially abundant serum metabolites and the 20 most abundant genera). b Heatmap of the correlations between the gut microbiome and clinical parameters. c Heatmap of the correlations between metabolites and clinical parameters
To explore the clinical significance of gut microbiome and metabolite dysregulation in patients with SCI, the putative correlations among gut microbiota abundance, metabolite abundance, and clinical parameters (including injury duration and neurological grade) were analyzed using Spearman’s correlation (Fig. 5b, c). We found that neurological grade was significantly and positively correlated with the abundance of Dorea (C = 0.655, p = 0.001), Faecalibacterium (C = 0.587, p = 0.005), Agathobacter (C = 0.567, p = 0.007), and Collinsella (C = 0.575, p = 0.006) and significantly and negatively correlated with the abundance of UBA1819 (C = − 0.709, p = 0.000). Simultaneously, neurological grade was identified as being significantly and positively correlated with the level of 12-hydroxydodecanoic acid (C = 0.455, p = 0.001), gamma-D-glutamylglycine (C = 0.712, p = 0.000), asparaginyl-hydroxyproline (C = 0.613, p = 0.004), and allocholic acid (C = 0.581, p = 0.007) and significantly and negatively correlated with the level of kojic acid (C = − 0.617, p = 0.004). In addition, injury duration showed a significant positive correlation with the abundance of UBA1819 (C = 0.660, p = 0.001) and a significant negative correlation with the abundance of Dorea (C = − 0.691, p = 0.001), Blautia (C = 0.575, p = 0.006), Faecalibacterium (C = − 0.618, p = 0.003), Agathobacter (C = 0.652, p = 0.001), and Collinsella (C = 0.646, p = 0.002). Meanwhile, injury duration was significantly and negatively correlated with the level of 2-methylbutyroylcarnitine (C = − 0.730, p = 0.000) and deoxycholic acid (C = 0.642, p = 0.002). The overall correlation results suggested that gut microorganisms UBA1819, Dorea, Faecalibacterium, Agathobacter, and Collinsella and the metabolites kojic acid, 12-hydroxydodecanoic acid, gamma-D-glutamylglycine, asparaginyl-hydroxyproline, and allocholic acid were associated with neurological grade. The results further suggested that the gut microbes UBA1819, Dorea, Blautia, Faecalibacterium, Agathobacter, and Collinsella and the metabolites 2-methylbutyroylcarnitine and deoxycholic acid were related to injury duration.
留言 (0)