
記住我










Ovarian cancer (OC) is a leading cause of cancer death among women worldwide, being the most lethal tumor of the female genital tract.1 Most women are diagnosed at advanced stages with a high risk of recurrence and poor outcomes.2 High-grade serous ovarian carcinoma (HGSOC) among all types of epithelial OC is the most frequent, representing approximately 70–80% of the cases. HGSOC is characterized by almost invariably TP53 mutations, genomic instability, and nearly half by defects in the DNA repair pathways.3
Despite an initial high response to platinum-based chemotherapy, nearly 70–80% will experience relapse and over time tumor cells will invariably become resistant.4 OC cells and their genomic landscape have been extensively studied, identifying potential targetable vulnerability leading to the development of new targeted therapies that include poly(ADP-ribose) polymerase inhibitors (PARPi). From early to advanced clinical trials, the activity of PARPi has been observed in patients with advanced OC, particularly for tumors with DNA repair pathway defects. Several PARPi are now used in clinical practice, particularly as maintenance after response to platinum-based chemotherapy in first-line setting. Targeted therapy is the cornerstone of precision medicine and has proved to be effective in selected patients.5
DNA Repair Pathways and Potential Vulnerability for Development of Therapeutic Strategies: PARP InhibitorsDNA damage can be induced by replicative stress (RS, DNA replication affected), enzymatic reactions, chemical modifications, radiation, and genotoxic agents.6 Among DNA lesions, single-strand breaks (SSB) are the most frequent and, if they are not properly repaired, they may result in double-strand breaks (DSB) that could affect transcription and block genome replication.7 The DNA damage response (DDR) system is constituted by a signaling network of pathways executed by specialized DNA repair proteins and factors which operate throughout the cell cycle.8 The effects of the DDR include cell cycle arrest, regulation of replication fork progression, and coordination of DNA repair; the latter minimizes DNA damage being shared to daughter cells (Figure 1).6 The response triggered by this system activates specific checkpoint pathways and their particular repair pathways. The SSB are usually repaired through excision repair mechanisms such as the single-strand break repair (SSBR), base excision repair (BER), and nucleotide excision repair (NER). Meanwhile, DSB is repaired by non-homologous end joining (NHEJ), homologous recombination repair (HRR), and alternative end joining (Figure 2). There are additional repair pathways such as mismatch repair and translesion synthesis.8 9
 Figure 1
Figure 1 DNA damage in ovarian cancer (OC). DNA damage is induced by different events in high-grade serous ovarian cancer (HGSOC). Once DNA is damaged, the DNA damage response (DDR) is triggered. The main effects of DDR are cell cycle arrest, regulation of the replication fork progression, and multiple DNA repair pathways. The response triggered by DDR activates specific checkpoint pathways and their particular repair pathways and factors. Major events in HGSOC include genomic instability and invariably loss of TP53. When TP53 is lost, the fidelity of the checkpoint G1/S is affected and then the cells rely on checkpoint G2/M, which make them more susceptible to specific targeted agents.3 6–9 Created with BioRender.com.
 Figure 2
Figure 2 Double-strand break (DSB) repair. Three main pathways help to restore DSB. Homologous recombination repair (HR) is the most important pathway which is mostly error free and high fidelity. It participates especially during phase G2 of the cell cycle and requires a homologous template to repair the DNA. Classical non-homologous end joining (NHEJ) is an error prone pathway and is more active in the G1 phase when DSB is being repaired. It does not require donor DNA contrary to what occurs in HR repair. The third pathway is Alternative NHEJ (aNHEJ), which comprises different sub-pathways including microhomology-mediated end joining (MMEJ) and is a major cause of genomic instability. This pathway functions independently of elements of the classic pathway; it acts as a back-up pathway when classic NHEJ or HR have failed.8 ,9–11 14 15 Created with Biorender.com.
PolyADP-ribose polymerases (PARPs) are one of the best known components of DDR. They are enzymes that catalyze the polymerization of ADP-ribose (PARylation), which is a post-translational modification of nuclear proteins induced by DNA damage. PARP1 promotes DNA repair and genome integrity, stabilization of DNA replication forks, and chromatin remodeling, and is the most studied member. PARP2 is also involved in DDR and its function is complementary to PARP1.6 9 10 The key role of PARP1 in DNA damage occurs early when there is a SSB, which is rapidly detected by the zinc-finger domain of PARP1 and uses NAD+ as a substrate releasing nicotinamide (PARylation). Linear and multibranched polymers of ADP-ribose are produced on target proteins or itself and favor cell survival.8 10 11 PARP1 then recruits downstream DNA repair factors (XRCC1, DNA ligase3, DNA polymerase theta)10 11 and contributes to the repair of DSB acting as a damage sensor. PARPs play a central role in detecting stalled replication forks and attract the MRE complex which is essential for end processing.9
If DNA damage cannot be repaired, apoptosis will occur; therefore DDR inhibition is an encouraging target including PARPi. When PARP is inhibited artificially, SSB lesions are not repaired and are converted to DSB. In the absence of BRCA, cells that are deficient in HRR will continue to accumulate DSB and lead to mitotic catastrophe and cell death, which eventually will result in a synthetic lethal relationship between PARP and BRCA.11–13 Synthetic lethality denotes the loss of cell viability secondary to the co-ocurrence of two alterations at the same time. In HGSOC, BRCA1 and BRCA2 act as tumor suppressors and are required for HRR to reduce genomic instability.8 12 13 BRCA1 starts the HRR pathway using a sister chromatid as a template for repair and recruits MRN complex, BRCA2 and RAD51.14 Additional evidence suggests that PARPi abolish a second pathway of the DDR, which in turn contributes to synthetic lethality in HR-deficient cells.11 Thus, high levels of genomic instability due to HRR loss make cells dependent on other pathways. Targeting this machinery is a promising strategy to exploit the vulnerability of defective DNA repair pathways9 11; both platinum agents and PARPi are genotoxic chemicals that exploit these DDR defects to induce cell death. The upregulation or downregulation of DDR proteins can drive sensitivity or resistance to these agents.14
PARPi can bind to the active site of PARP inhibiting its catalytic activity and trapping the complex PARP-DNA. This may additionally cause obstruction to replication forks, which require BRCA-dependent HRR to be resolved.13 The efficiency and potency for trapping is variable among the PARPi and it is associated with cytotoxicity magnitude. Of the available PARPi, talazoparib is the most effective trapper; meanwhile olaparib, rucaparib and niraparib trap PARP 100-fold more than veliparib.11 15 From pre-clinical to early clinical trials, PARPi have demonstrated anti-tumor activity, particularly in BRCA deficiency and, more broadly, in tumors with HR deficiency. Further investigations have reported significant benefit of PARPi in the context of maintenance therapy after platinum-based chemotherapy in HGSOC; currently, olaparib and niraparib are approved for first-line maintenance by the US Food and Drug Administration16–36 (online supplemental table 1).
Table 1Relevant clinical trials of PARPi in OC
Defining Resistance to PARP Inhibitors: Identification of Mechanisms of ResistanceDespite the activity of PARPi, the majority of patients will ultimately recur. One approach to determine resistance may be based on timing from cancer recurrence to last dose of treatment, as it was clinically defined for platinum sensitivity or resistance and used to outline the benefit of platinum rechallenge. This concept of PARPi maintenance re-challenge after PARPi exposure has been recently tested in the phase III trial OReO, which investigated the efficacy of olaparib maintenance in patients with relapsed OC who have had progression following a specific duration of maintenance with PARPi. They included two patient cohorts (n=220) after response to platinum-based chemotherapy: (1) patients with BRCA mutated (BRCAm) OC who had prior PARPi exposure for at least 18 months after first-line or >12 months after second or later chemotherapy; and (2) non-BRCAm patients who had prior PARPi exposure for >12 months after frontline or >6 months after subsequent lines of chemotherapy. The study showed that patients who received PARPi again had longer progression-free survival (PFS) compared with placebo regardless of BRCA mutational status (BRCAm cohort: 4.3 vs 2.8 months (HR 0.57 (95% CI 0.37 to 0.87); p=0.022); non-BRCAm cohort: 5.3 vs 2.8 months (HR 0.43 (95% CI 0.26 to 0.71); p=0.0023)).37 Of note, most patients enrolled in this study received the initial PARPi maintenance at the time of relapse and progressed on PARPi at the time of the OReO enrolment; yet most recurrences will now occur after discontinuation of PARPi in the first line and there is a lack of data on rechallenge PARPi in this setting. However, this study highlights that PARPi exposure is not equivalent to PARPi resistance. It is noteworthy that, when analyzing the PFS Kaplan–Meier curves, there is a sub-set of patients who definitively benefit from PARPi rechallenge while others do not have benefit at all (early resistance). In line with this, a sub-group analysis of 37 patients treated with niraparib in the fourth or more therapy line after prior PARPi therapy had clinical benefit at 16 weeks of 20% in the QUADRA trial, indicating potential disease control when rechallenging patients with PARPi.38 Translational data are needed to determine the rationale of this strategy in a particular sub-set of patients who would derive benefit from rechallenging after PARPi exposure. Indeed, BRCAm OC cells can develop mechanisms to adapt and resist under selective PARPi pressure, driving the emergence of resistant clones.39 Evolutionary pressure can promote changes in the tumor microenvironment and clonal heterogeneity (functional and genomic). These resistant clones will exhibit more robust mechanisms of resistance over time.40 The concept of PARPi resistance is an evolving concept and there is no established treatment-free interval defining sensitivity/resistance to PARPi. Patients may fail PARPi from the beginning of treatment because they have a competent DSB repair mechanism or pre-existing resistance mechanisms usually related to platinum resistance (primary resistance) or patients may progress during the course of treatment with PARPi (secondary resistance).
Sensitivity to platinum-based chemotherapy in HGSOC may represent a clinical predictive marker for PARPi response since both target DNA repair,41–44 yet acquired resistance to these drugs does not develop entirely in parallel.14 PARPi and platinum agents may share common and overlapping resistance alterations in the HR pathway, especially those related to secondary BRCA reversion mutations, loss of 53BP1, and replication fork protection.45 However, some mechanisms may be distinct based on their specific target or drug delivery, as platinum resistance is associated with defects in the NER pathway and copper efflux transporters while PARPi have alterations in the MDR1/ABCB1 drug efflux pump. Platinum agents form mono-adducts in the DNA resulting in changes to the DNA double helix and leading to accumulation of DSB similar to PARPi and, as such, specific mechanisms of platinum and PARPi resistance partially overlap, with DDR playing a central role.14 In two randomized trials HR gene reversion mutations were detected pre-PARPi but after platinum-based chemotherapy, predicting primary resistance to PARPi. The ARIEL-4 study included patients with BRCAm relapsed OC who received rucaparib versus standard of care (SoC) chemotherapy. Before treatment, 7% of patients (23/349) had BRCA reversion mutations (of whom 17 had platinum-resistant and six had partially platinum-sensitive disease) and their median PFS was shorter with rucaparib (2.9 vs 5.5 months, HR 2.769 (95% CI 0.989 to 7.755)), predicting primary resistance to rucaparib.28 In the SOLO-3 study, six heavily pre-treated patients with platinum-sensitive BRCAm OC presented BRCA2 reversion mutations at baseline. Among them, there were no responses with olaparib.46 In addition, according to retrospective studies, the benefit of platinum agents after progression on PARPi, which are part of the SoC in this context, may depend on the platinum-free interval.41 43 Post hoc analysis from the SOLO-2 trial showed that patients treated with chemotherapy following progression on placebo had a significantly longer time to second progression compared with patients who progressed on PARPi (12.1 vs 6.9 months; HR 2.17 (95% CI 1.47 to 3.19)); this effect was more pronounced in patients with BRCA1. The patients who received platinum agents post-progression on SOLO-2 had poorer outcomes if they were previously treated with PARPi, but there were no significant differences between the two groups if they received non-platinum regimens as next line therapy. Further prospective evidence is needed to identify mechanisms of resistance in a timely fashion.42
Translational research has described the main mechanisms underlying acquired resistance to PARPi; however, they are still not fully elucidated and remain a challenge in the clinical setting since one or several of these mechanisms may be present in the same individual and evolve over time.45 47 The resistance mechanisms include restoration of HR, drug/target-related resistance, and restoration of replication forks15 47 (see online supplemental table 2 and Figure 3). Additional investigations have also identified other mechanisms leading to a better understanding of the molecular basis that mediates the resistance.11 Nonetheless, their clinical relevance has not been evaluated systematically.48
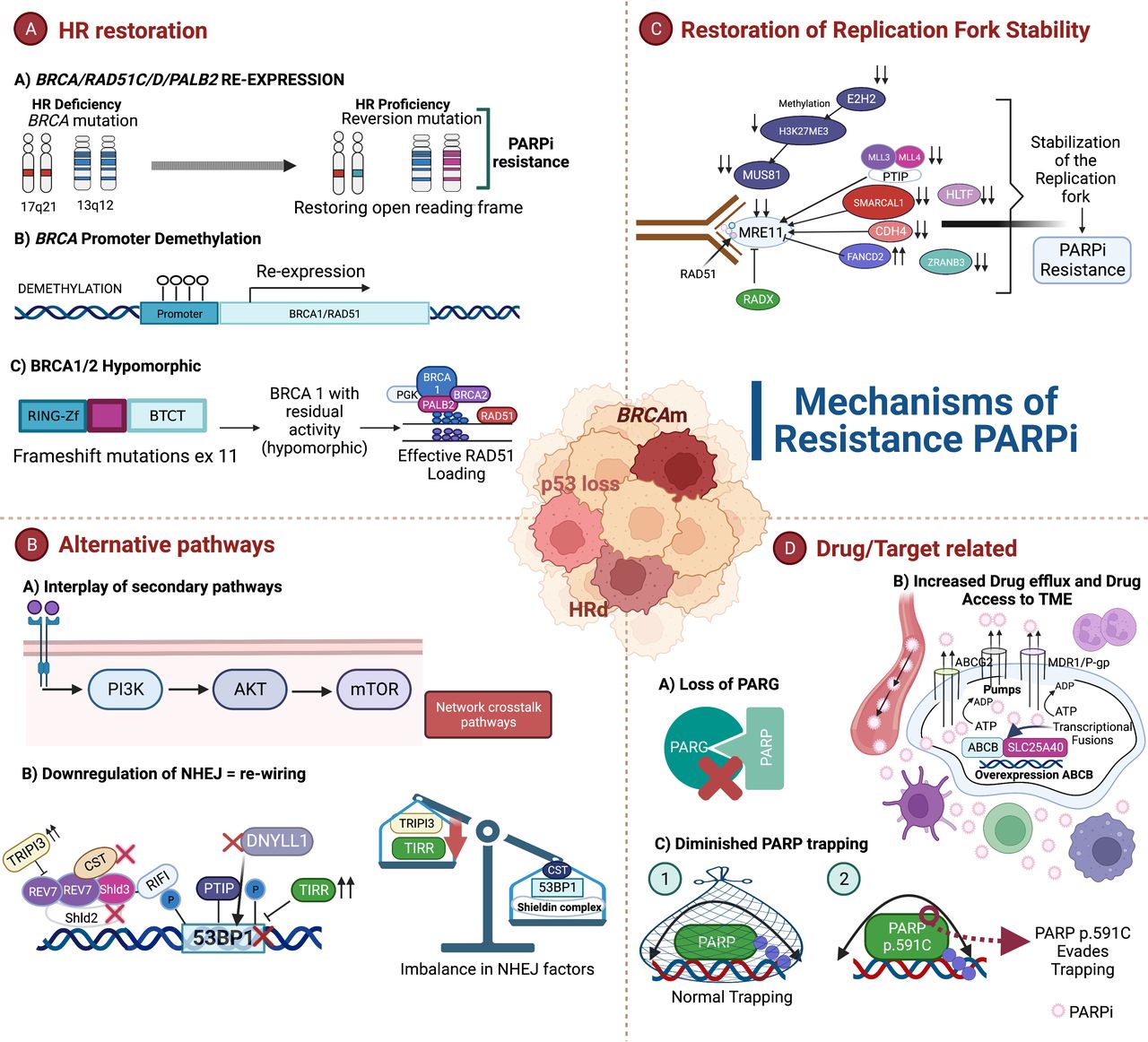 Figure 3
Figure 3 Mechanisms of PARPi resistance. (A) The most studied mechanism of resistance is the homologous recombination (HR) restoration by genetic and epigenetic changes: (a) re-expression of the genes BRCA/RAD51C/RAD51D/PALB2 is achieved through reversion mutations; (b) intrachromosomal genomic rearrangements lead to the re-expression of BRCA by the demethylation of a heterologous promoter; and (c) BRCA hypomorphs have residual activity and are able to confer incomplete resistance to PARPi.11 47–52 (B) Alternative pathways are involved in the development of resistance to PARPi: (a) The crosstalk between secondary pathways with HR plays a crucial role to induce resistance and are attractive therapeutic targets to try to overcome resistance; and (b) downregulation of non-homologous end joining (NHEJ).15 54–56 80 81 (C) Loss of different factors induce stabilization of the replication fork leading to PARPi resistance.65 66 (D) Drug/target-related mechanisms include: (a) the loss of PARG;9 59 61 (b) an increased drug efflux (overexpression of ABCB) and drug access to tumor microenvironment (TME) can also lead to resistance;57–60 and (c) limited PARP trapping has been detected in PARPi-resistant ovarian cancer cells.59 63 64 Created with Biorender.com.
Restoration of homologous restoration (HR)HR recovery is considered the main mechanism of resistance to PARPi and platinum agents that is achieved through restoration of proteins involved in this process (BRCA-dependent) and by altering specific factors of DNA repair pathways (BRCA-independent).11 47
Secondary reversion mutations of key HR genes are a well described mechanism of resistance and clinically confirmed. The reversion mutations re-establish functional protein expression and the ability of cancer cells to repair DNA by HRR; most of them happen when the open reading frame is restored.4 49 50 Ter Brugge and colleagues found that specific de novo intrachromosomal genomic rearrangements induced the re-expression of BRCA1 as well. These genomic alterations lead BRCA1 transcription under the control of a heterologous promoter; BRCA1/2 re-expression in this scenario is driven by genetic and epigenetic changes.50 An additional mechanism that allows regaining functional HR proteins is the BRCA hypomorphs.49 51 Of note, frameshift mutations in exon 11 may generate a hypomorphic isoform BRCA1 (BRCA1-delta11q) leading to incomplete resistance.51 Reversion mutations can occur in other HR genes including RAD51C, RAD51D, and PALB2.52 Specifically, RAD51 participates in the early steps of HRR and protection of the fork. Pre-clinical findings have suggested that RAD51 alterations including reversion mutations and loss of RAD51C promoter methylation are involved in the recovery of HRR. RAD51C methylation can be lost under PARPi treatment pressure similar to BRCA1 promoter methylation.53
Restoration of HR can also result from rewiring of DDR in BRCA-deficient cells, achieved by downregulating NHEJ.15 54 The absence of NHEJ proteins including 53BP1, RIF1, and the Shieldin complex rescues embryonic lethality and decreases chromosomal instability.15 Meanwhile, the upregulation of TRIP13, which disables the Shieldin complex, and TIRR, which masks 53BP1, confer PARPi resistance.15 55 56 Additional loss of DYNLL1, which blocks MRE11, and the CST complex, which blocks RAD51 loading, can lead to the restoration of end-resection when BRCA is deficient.15 All of the above factors, if they are altered, produce increased resection in BRCA1-deficient cells and therefore favor PARPi resistance.
Drug/Target-Related ResistanceThe EVOLVE study looked at patients who progressed on PARPi and were rechallenged with PARPi and cediranib. The analyses of archival tumor and baseline biopsies at the time of PARPi progression identified PARPi resistance mechanisms. Reversion mutations in HR genes were the most frequent mechanism, followed by CCNE1 amplification and upregulation of drug efflux pump gene ABCB.48 The latter can translate into dysregulation in the cellular availability of PARPi. Different models have shown that there is an over-expression of drug-efflux transporter genes in tumors that displayed resistance to PARPi.57 Data have revealed that ovarian tumor cells that were resistant to olaparib showed no cross-resistance to veliparib,58 which in turn does not exclude sensitivity to alternative PARPi in the scenario of P-glycoprotein-mediated drug resistance.59 A recent investigation suggests that the overexpression of ABCB could be mediated by chromosomal translocations and occur as a consequence of the control of a strong promoter (SLC25A40) while maintaining its open reading frame intact.60
In addition, an abnormal PARP1 and the loss of PARG can hinder PARP1 trapping leading to resistance. Once PAR is generated during the DNA reparation, its rapid turnover is also required so DNA repair is not affected. The most extensively studied degrading enzyme is the PARG.9 PARPi decreases PARP1 activity, and the residual activity is decreased further by PARG. When PARG is lost, PARP1 signaling may be re-established,59 and may abolish PARP1 trapping on DNA by preventing its binding. This is in line with pre-clinical evidence which showed that depletion of PARG is correlated with PARPi resistance by restoring PAR formation and partially rescuing PARP1 signaling.61 This is an HR-independent mechanism in which the selective loss of PARG activity is sufficient to restore PARylation,59 slow down replication forks, and prevent fork restart by RECQ1 inhibition.62 Functional PARP is also necessary for the action of PARPi that requires trapping of PARP at damaged DNA to produce cytotoxicity. PARPi can trap PARP1 and PARP2 on DNA, and PARP1 depletion is associated with reduced sensitivity to PARPi because it is the dominant target for DNA trapping.63 In BRCAm tumor cells, induction of mutations in the genes encoding PARP1 is associated with altered PARP trapping and resistance to PARPi.59 64 A specific non-synonymous substitution (p.R591C) in PARP1 has been detected in PARPi-resistant OC cells and is associated with a decrease in PARP trapping on DNA.64
Stabilization of the Replication ForkStabilization of the replication fork is an alternative mechanism that induces PARPi resistance. In DNA damage, PARP1 and BRCA participate in preventing DNA at stalled replication forks to be degraded by nucleases MRE11 and MUS81, which are recruited by PTIP and EZH2. PARPi leads to the accumulation of SSB and replication forks are stalled, nucleases are then recruited, which in turn induce replication fork collapse and DSB. The activity of EZH2 and PTIP is limited in BRCA-deficient cells, reducing the recruitment of the nucleases and favoring DNA protection from extensive degradation. Loss of chromatin remodeler factors SMARCAL1, ZRANB3 and HLTF, RADX deletion, and silencing of Schlafen 11 by methylation in BRCA-deficient cells promotes fork protection leading to PARPi resistance.65 66
Other Mechanisms of ResistanceThe activation and operation of other oncogenic pathways including PI3K/AKT/mTOR and RAF/MEK have been described as potential mechanisms for resistance to PARPi due to the PARP-PI3K-AKT crosstalk with HRR. Pre-clinical evidence showed that PI3K-AKT pathway is activated by PARPi inducing apoptosis resistance and mitochondrial protection that limit cytostatic efficacy of PARPi.67 The cell cycle checkpoint signaling is also crucial in regulating DDR and is a key participant in some of the mechanisms of resistance by promoting repair of DNA lesions and proliferation.68 The identification and description of each of these factors involved in these pathways have allowed the creation of therapeutic targets to overcome resistance.
Detection of Mechanisms of Resistance to PARP Inhibitors in the ClinicIn the clinical setting, secondary BRCA1/2 reversion mutations have been identified in tumor biopsies and cell-free DNA (cfDNA) of patients with OC and their implication in the treatment has been studied.4 48 49 52 69 A comprehensive analysis of reversion mutations included 327 BRCAm patients who progressed on PARPi and platinum-based chemotherapy, of whom 234 had OC. Among the patients with OC, those with BRCA1 had 21.4% reversion mutations while those with BRCA2 had 27.5%. The majority of reversion mutations were deletions followed by insertions and were able to recover the BRCA activity by restoring the open reading frame.49 Additional research is needed to assess the impact of the primary mutation location and potential risk for mutation reversion. Determined regions may be less permissive for amino acid changes in BRCA1/2 than others. As such, the nature of the primary mutation and location may be helpful to determine the repair event that will lead to acquisition of secondary mutations.50 Recently, cfDNA was analyzed in over 500 plasma samples collected at baseline and at progression in patients with BRCAm OC and breast cancer treated with chemotherapy or olaparib from the phase II/III studies LIGHT, SOLO-3, and OlympiAD. BRCA reversion mutations were detected in 3.5% and 4.5% of patients with breast cancer and OC at baseline while, at progression, 43% and 25% of patients who received olaparib and 9% and 3% of patients on chemotherapy with breast cancer and OC, respectively, had BRCA reversion mutations. Reversion mutations in BRCA1 were mainly in exon 11, although the specific domain was not identified, while BRCA2 mutations were seen in the three pre-defined regions (exon 11, N- and C-terminus). The reversion mutations were found as single or multiple, suggesting multiple events within the same tumor, and the location and type of the mutation also reflected the functional importance of BRCA protein domains.70
The parallel between PARPi and platinum resistance is also highlighted in the ARIEL-4 study. BRCAm relapsed HGSOC patients were randomized to receive rucaparib or chemotherapy; the election of the regimen depended on the platinum-free interval (weekly paclitaxel or platinum-based chemotherapy). CfDNA was collected at baseline and after progression. Cross-over was allowed and patients initially assigned to weekly paclitaxel were allowed to receive rucaparib post progression. In this small sub-group of patients, the median duration of rucaparib was higher than expected. Translation analysis showed a decrease in BRCA reversion mutations in three of four paclitaxel-treated patients, which may in part explain the response to rucaparib, but the exact mechanism is to be determined.29
A retrospective analysis performed by Hu and colleagues explored the mutation profiles associated with PARPi resistance using sequential cfDNA sampling in patients with platinum-sensitive OC. Patients who had germline pathogenic mutations and new somatic MRE11A mutations in post-treatment with olaparib had a poor prognosis compared with non-carriers; these mutations were mainly concentrated in MRE11A:p.K464R, which could be an indicator for resistance monitoring of olaparib.71 Identifying potential biomarkers based on the mechanisms of resistance could lead to the development of models that allow selection of patients. However, the central challenge remains in how to incorporate all the findings into clinical practice. Currently there is a lack of a cohesive model integrating clinical, histologic, and genomic data into the management that may help to develop specific strategies to overcome resistance. Refining the algorithms of treatment with the identification and reproducibility of biomarkers of resistance is important for personalized/precision medicine in OC.
Novel Therapeutic Strategies for the Post-PARPi EraOngoing research in the era of PARPi in OC is considering the following clinical scenarios: (1) preventing resistance (pre-PARPi) by decreasing the risk of developing mechanisms of resistance and particularly trying to identify the patients at higher risk; (2) overcoming resistance (post-PARPi) by targeting the specific mechanisms of resistance with novel therapies; and (3) bypassing resistance (post-PARPi) by suppressing alternative pathways or improving drug delivery.
Several agents are being studied in this context; however, there are currently no approved therapies alone or in combination with PARPi/chemotherapy in patients whose disease progresses on PARPi (Table 1). The capability to restore sensitivity to PARPi is an encouraging line of research which considers the dynamic and diverse genetic landscape in HGSOC (Figure 4). Understanding the underlying mechanisms of resistance and recognizing the potential of heterogeneous clones will provide relevant information to guide the optimal way for individualized therapy.40
Table 1Summary of selected clinical trials that evaluate new promising agents for patients who progressed on PARPi
 Figure 4
Figure 4 Novel therapeutic strategies for the post-PARPi era. Multiple complex cellular processes are involved in the DNA damage response (DDR) system and interplay with other secondary pathways. PI3K/AKT is a frequent pathway altered in high-grade serous ovarian cancer (HGSOC). The pathway becomes activated in response to external signals that will lead to the activation of the complex mammalian target of rapamycin (mTOR). It can lead to the increase of BRCA1, RAD50, RAD51 and FANC2. In addition, the RAF/MEK pathway is activated and participates in the regulation of the transcription of BRCA1.67 80 85–87 AKT can also be activated by ATM (ataxia-telangiectasia mutant), ATR (ataxia-telangiectasia and Rad3 related) and DNA-PK (DNA-dependent protein kinase) showing the diverse cellular functions in which AKT participates.80 82 85 The ATM/ATR pathways are also involved very closely with PARPi resistance. In HGSOC, high levels of replicative stress trigger the DDR which promote cell cycle arrest. When the DNA is damaged, it will depend on the type of lesion to activate a specific pathway. In the case of double-strand breaks, ATM is activated, which enables CHK2 activity. P53 is stabilized by CHK2 and maintains the cell cycle arrest. When a single-strand break is present, ATR is activated and phosphorylates CHK1, which in turn prevents G1/S and G2/M transition leading to cell cycle arrest and apoptosis. CHK1 activates WEE1-like protein kinase (WEE1), which is a negative regulator of the CDK preventing the transition between phases of the cell cycle. Myelin Transcription Factor 1 (MYT1) is also a negative regulator of CDK1 and possess similar functions to WEE1.14 63 68 80 81 Different inhibitors have been developed to try to overcome PARPi resistance based on this complex network of pathways. Created with Biorender.com.
Targeting RS: Cell Cycle Checkpoint Proteins InhibitionIn HGSOC, many of the defining molecular abnormalities impact DNA repair and the cell cycle. The raised levels of RS trigger the DDR sensors such as ataxia telangiectasia and Rad3 related kinase (ATR), ataxia telangiectasia mutated (ATM), checkpoint kinase 1 (CHK1), WEE1-like protein kinase (WEE1), and membrane-associated tyrosine- and threonine-specific CDC2-inhibitory kinase (MYT1), which promote cell cycle arrest in specific phases while restoring stalled replication forks allowing cell reparation and thus survival. Ovarian tumor cells that are resistant to PARPi exhibit enhanced dependency on other DNA repair pathways and cell cycle mechanisms leading to a wide therapeutic window of opportunity.68 72 Targeting cell cycle checkpoints is of particular interest given the potential impact on inducing synthetic lethality among TP53 mutant tumors. As TP53 is almost ubiquitously lost in HGSOC (genomic unstable tumor), it has high levels of oncogenic stress. TP53 mutation induces the loss of G1/S checkpoint fidelity and eventual cell cycle arrest and apoptosis, leading the tumor cells to mostly depend on S/G2 checkpoint. ATR is a well-known regulator of cell death and controls cell cycle arrest from S to G2 phases. In cells with TP53 mutation, ATR promotes checkpoint-defective cells and the inhibition of this target might become lethal.68
Early clinical trials have shown the potential activity of different ATR inhibitors (ATRi). A phase I trial reported that BAY 1895344 with dose >40 mg twice daily was well tolerated and showed anti-tumor activity in 21 patients with heavily pre-treated advanced solid tumors with an objective response rate (ORR) of 30.7% (4/13). All the responders had ATM loss or ATM mutation and one patient with BRCA1m HGSOC which was olaparib- and chemotherapy-resistant had a durable response.73 Elimusertib showed clinical activity as well in patients with advanced solid tumors resistant to standard treatments with DDR defects, of which 45 had gynecological tumors. A durable clinical benefit lasting >6 months in 27.8% of patients with OC was achieved, including those resistant to platinum-based chemotherapy and those with prior PARPi. However, significant grade 3–4 hematological toxicity was reported.74 In addition, RP-3500 was evaluated in the phase I TRESR trial. Twenty patients with DDR-deficient OC were included, 18 of whom had progressed on prior PARPi and 17 were platinum resistant. The ORR was 25% and median PFS was 35 weeks. Two patients with post-PARPi BRCA1 reversion mutations had treatment durations of 17 and 29 weeks.75
The combination of PARPi and ATRi has shown a synergistic effect that leads to DNA damage and durable tumor regression. The CAPRI trial evaluated the combination of olaparib and ceralasertib in platinum-sensitive HR-deficient HGSOC which progressed on prior PARPi and had a clinical benefit from them (>12 months for first line and >6 months for >second line). The ORR was 46% (n=6) and median PFS was 7.5 months. The combination showed encouraging clinical activity and acceptable toxicity.76 This combination was assessed as well in the OLAPCO trial reporting an ORR of 14% (1/7) and clinical benefit of 86% (6/7) in seven patients with PARPi-resistant BRCAm HGSOC.77 Berzosertib given with gemcitabine (nucleoside metabolic inhibitor that induces additional RS leading to mitotic catastrophe) showed clinical activity in platinum-resistant OC. Of the 70 patients included, 19% and 32% had prior PARPi in the combination and single agent groups, respectively. The median PFS was 22.9 weeks in the combination group and 14.7 weeks in the gemcitabine group (HR 0.57 (95% CI 0.33 to 0.98); p=0.04).78 Interestingly, they analyzed the level of RS according to a pre-established score (high if at least one RS alteration was present versus low if none); only those with low RS had a PFS benefit by adding berzosertib to gemcitabine (HR 0.34 (90% CI 0.13 to 0.86)), suggesting a potential role as biomarker and worthy of future evaluations.79
CHK1/2 also participate in the cell cycle by inhibiting cell division at S and G2/M checkpoints and allowing time to repair DNA. Tumor cells with TP53 mutation rely on these checkpoints, suggesting that there could be a benefit by CHK inhibition that could prevent progression and tumor growth.14 The role of the CHK1/2 inhibitor prexasertib in HGSOC has been explored in combination with olaparib in a phase I trial which demonstrated some anti-tumoral activity (4/18 patients with BRCA1-mutant, PARPi-resistant, HGSOC achieved partial responses) and could mediate HR by decreasing RAD51 foci. The safety profile was acceptable, hematological toxicity being the most frequent.80
The WEE1 kinase family consists of three serine/threonine kinases (WEE1, PKMYT1 and WEE1B) that regulate the cell cycle checkpoint at G2/M, which favors apoptosis when there is DNA damage.81 The inhibition of WEE1 induces anti-tumor activity in TP53 mutant cancer cells as they depend on this checkpoint.72 Adavosertib as a single agent or in combination with olaparib in OC following PARPi progression (previous benefit from PARPi not required) produced an ORR and median PFS of 23% and 5.5 months and 29% and 6.8 months, respectively, in 70 evaluable patients in the preliminary results of the phase II EFFORT. Grade 3 and 4 toxicities were observed in both groups and were manageable; however, 56% in the combination arm required dose reduction and 85% had a dose interruption.82 Modulating the cell cycle checkpoint signaling in OC with agents targeting DDR pathways (ATR-CHK1-WEE1 pathway) is an exciting option with mechanistic rationale for the combination with PARPi or cytotoxic chemotherapy as they have the potential to induce synthetic lethality. All these combinations require comprehensive pre-clinical data to assess schedule timing of dosing for optimal strategy combination and to limit the induced hematological toxicities.
DNA polymerase thetaDNA polymerase theta (POLθ) has recently been studied as a new promising target. HR-deficient tumor cells become dependent on MMEJ. POLθ is then upregulated in OC and compensates the loss of HR through mediating DSB repair (MMEJ). In mouse and PDX models, the inhibition of POLθ by the antibiotic novobiocin and allosteric inhibitor ART558 in HR- and BRCA-deficient tumor cells elicit DNA damage, synthetic lethality, and enhance the activity of PARPi.83 84 Specifically, concurrent defects in 53BP1 and Shieldin complex and BRCA deficiency cause PARPi resistance but result in profound sensitivity to ART558.84
Secondary Pathways Crosstalk: PI3K/AKT InhibitionPI3K signaling is a common altered signaling in OC and can regulate HRR via mTOR-mediated and nuclear PI3K can regulate HRR and NHEJ repair via AKT. Once PI3K is activated, mTORC1 increases the proteins of the HR pathway including BRCA1, RAD50 and RAD51, while mTORC2 also produce high levels of FANCD2. Moreover, PI3K indirectly regulate the transcription of BRCA through the activation of MEK/ERK/EST1 and in an independent fashion ATM induce nuclear AKT activation in response to DSB (complex ATM-NEMO).85 Defects in BRCA might increase the activation of the PI3K/AKT pathway and the inhibition of PI3K produces disruption of HR.86 87 Consideration of combining PARPi and PI3K/AKT inhibitors is currently being evaluated in clinical trials. Two phase I trials showed manageable toxicity with the combination olaparib and capivasertib; one of them included 13 patients with OC previously exposed to PARPi, five of whom had clinical benefit and three had a duration of response between 31 and 115 weeks.88 89
Improving Drug Delivery: Antibody–Drug Conjugates (ADC)An important alternative to this strategy is trying to improve drug delivery by the creation of ADC that are promising agents designed to target specific antigens of ovarian tumor cells with direct delivery of cytotoxic agents to overcome resistant disease while limiting toxicity.90 There are many ADC under investigation including mirvetuximab, which has been explored in phase II and III trials showing clinical activity with a favorable safety profile in heavily pre-treated OC and high levels of folate receptor alpha (FRα). Early studies have evaluated the combination of this ADC with other agents reporting anti-tumor activity in patients with prior PARPi exposure. A phase I study evaluated mirvetuximab and gemcitabine in OC, breast, and endometrial cancer. In the cohort of 30 patients with recurrent FRα-positive platinum-resistant OC (53% had prior PARPi), the ORR was 36.7% with a median PFS of 5.4 months.91 A second phase I trial enrolled five patients with recurrent endometrial cancer and 13 with OC, of which five had received prior PARPi; the ORR achieved with mirvetuximab and rucaparib was 40% and median PFS of 12.6 months in those with OC.92 Pivotal information from the SORAYA trial reported an ORR of 32.4% in 105 patients, with a median duration of response (DOR) of 6.9 months and PFS of 4.3 months. The sub-group analysis looked at whether patients who had received prior PARPi (n=51) had benefit. They had an ORR of 38% and median DOR of 6.9 months.93 The role of the ADCs in overcoming resistance to PARPi is a promising strategy and forthcoming studies will explore new target antigens leading to a new generation of ADC.
New Generation PARPi: Avoiding Drug-Efflux PumpsAs drug-efflux pumps can be overexpressed in PARPi-resistant tumor cells, the creation of highly selective PARPi could help to avoid this mechanism (not a substrate for MDR-1 mediated efflux) and overcome resistance. A next-generation PARPi (AZD5305) has been developed and targets specifically PARP1. It showed promising clinical activity in different types of HR-deficient cancer including OC, according to the phase I/IIa PETRA trial. The ORR was 28% (7/25) including platinum- and PARPi-resistant patients. This novel PARPi achieved a higher above-target effective concentration compared with first-generation PARPi and was well tolerated with lower rates of hematological toxicity. Being highly selective and less toxic may enable broader combination options.94
Other Therapies Currently under InvestigationPARPi may modulate the tumor micro-environment, increase the tumor mutational load and neoantigen expression, which may be a strategy to enhance immunotherapy responsiveness.95 A phase II trial enrolled 35 patients with recurrent heavily pre-treated OC who received olaparib and durvalumab. Most of them were platinum-resistant (86%, 30/35) and two who did not have BRCA reversion mutations at baseline had received prior PARPi. The ORR was 14% (5/35) and the median PFS was 3.9 months, but it did not meet the pre-specified primary endpoint.96 Adding PD-L1/PD-1 therapy has been assessed earlier in the disease trajectory in large phase III first-line trials, for which results are awaited, including the triplet combination with PARPi, PD-L1/PD-1 therapy and antiangiogenics. Diverse combinations are currently under investigation to try to overcome resistance to PARPi (Table 2). Multiple mechanisms co-exist and some may not have been identified yet, highlighting the importance of biomarker analyses as part of clinical trials.
Table 2Ongoing clinical trials evaluating therapies in patients with OC who progressed on PARPi or had prior exposure
ConclusionsThe pre-clinical models, in vivo studies, and clinical trials available so far provide highly relevant data about novel therapeutic strategies and are helping to elucidate the multiple mechanisms involved in the PARPi resistance in OC. This is a unique setting that involves changes in the genes/proteins that participates in the DNA repair pathways and also affects kinases involved in the checkpoint cell cycle and tumor microenvironment. The heterogeneity of these mechanisms may prevail even in one patient, which represents a challenge yet an opportunity to develop novel
留言 (0)