
記住我







The year 2022 marked the 50th anniversary of the groundbreaking collaboration between Michael S. Brown and Joseph L. Goldstein, who established their joint laboratory at the University of Texas Southwestern in 1972. Among their many contributions to science and medicine, they discovered the low-density lipoprotein (LDL) receptor, described receptor-mediated endocytosis, and elucidated the first molecular understanding of familial hypercholesterolemia (FH) [1]. This foundational work provided a direct connection between genetic inheritance and atherosclerotic risk mediated through LDL particles in the blood. The subsequent decades of research have revealed FH to be a truly complex disease [2]. New questions have arisen about the relationships between genotype, phenotype, and risk, and the precise definition of FH itself is a topic of some contention [3]. Central to our understanding of the disorder is the question, ‘Does LDL fully explain atherosclerotic risk in FH?’
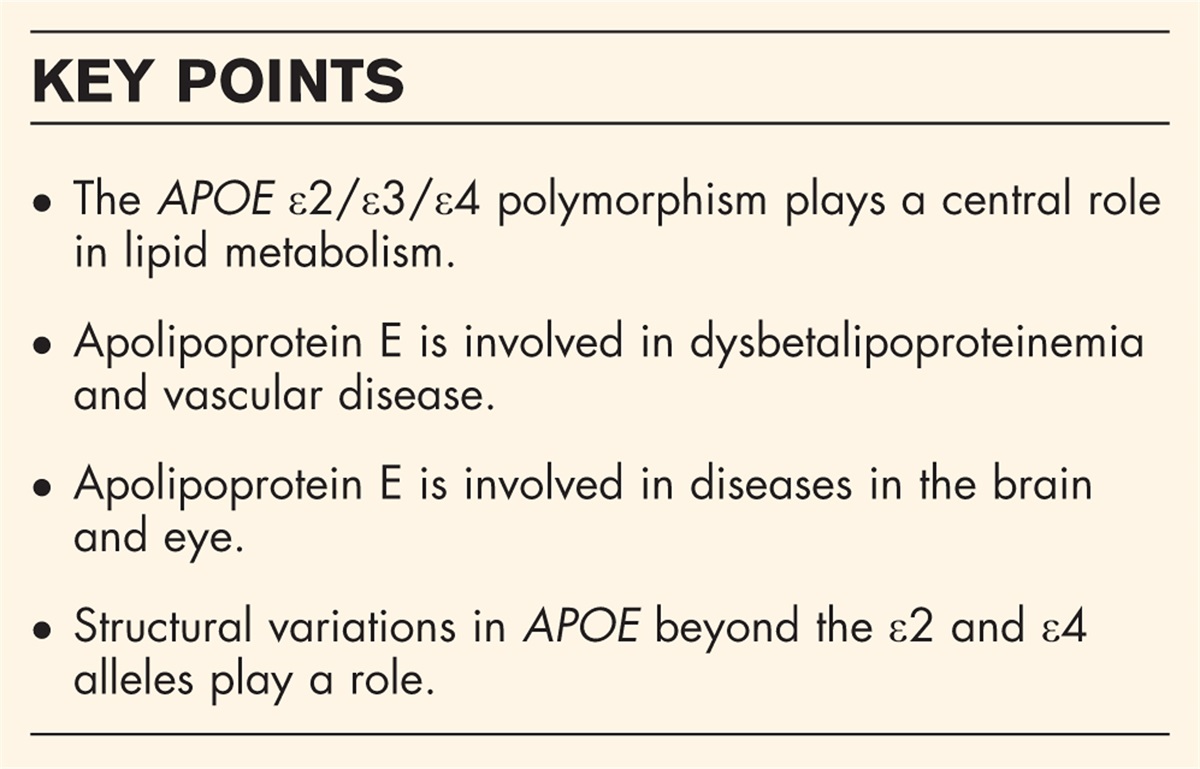
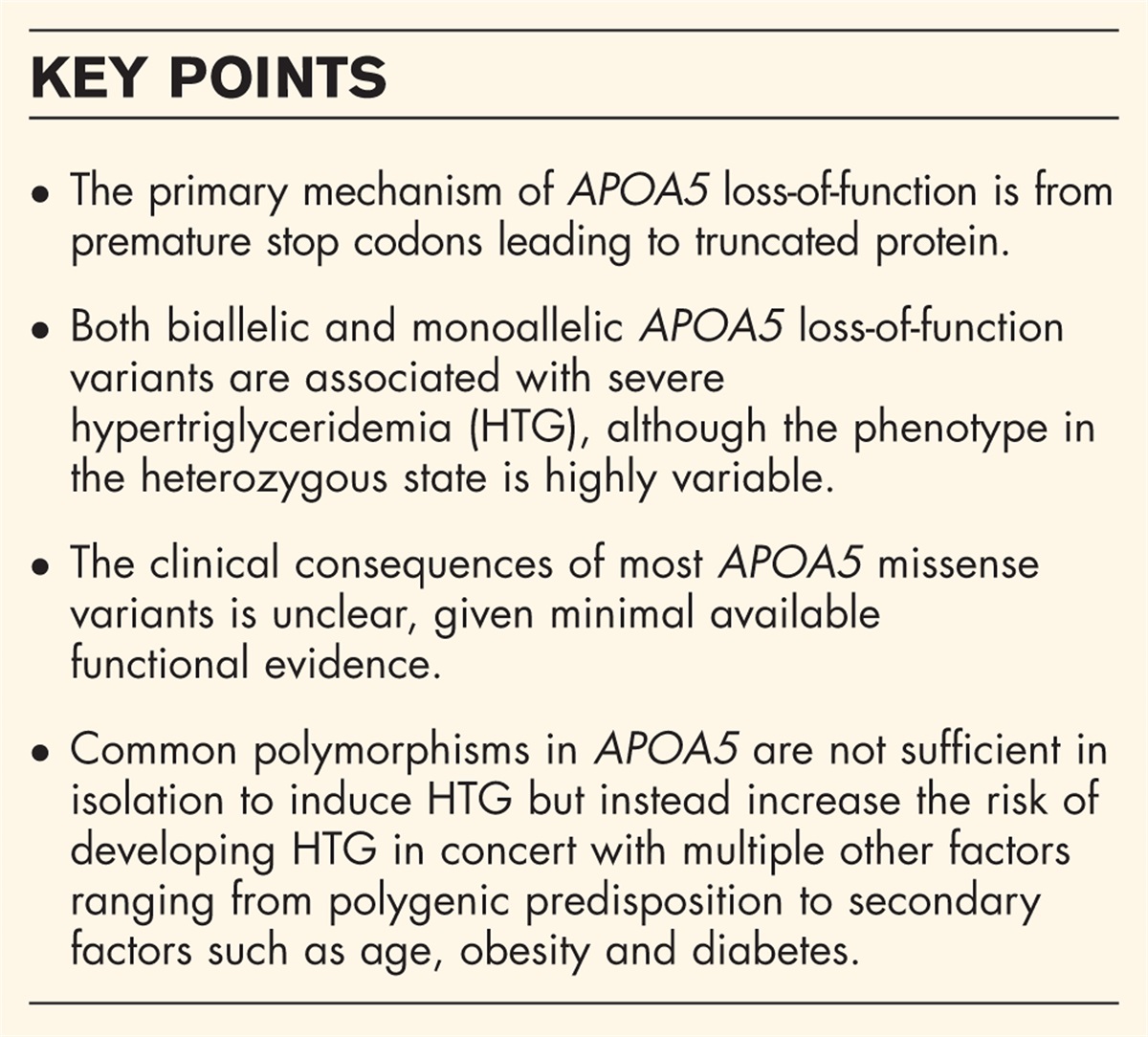
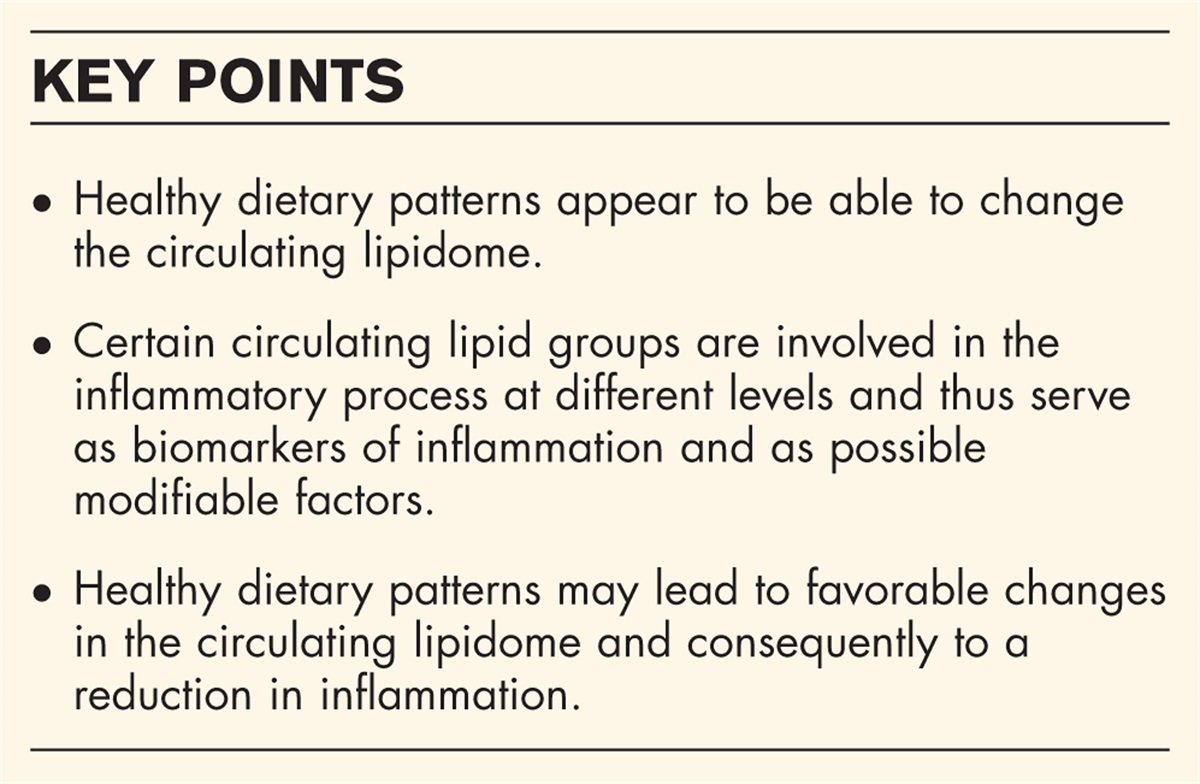
 Box 1:
Box 1: no caption available
MAIN FH variantsIn the early 20th century, ‘essential familial hypercholesterolemia’ was first described as an autosomal dominant genetic condition associated with elevated blood cholesterol, physical findings of xanthoma tuberosum and xanthelasmas, and coronary heart disease [4–6]. Of course, these early observations were made long prior to any molecular understanding of FH. Instead, researchers relied on careful phenotyping and pedigree analyses. Brown and Goldstein identified disruption of the LDL receptor gene (LDLR) as causative for FH, but this simple ‘one gene one disease’ model did not last long. Before the end of the century, two additional loci were implicated in severe autosomal dominant hypercholesterolemia [7,8]. The first was APOB (which encodes apolipoprotein B100). The second was subsequently identified as PCSK9 (which encodes proprotein convertase subtilisin/kexin type 9) [9]. Pathogenic rare variants at these three loci - LDLR, APOB, and PCSK9 - account for the majority of genetically diagnosed FH and have been the primary focus of population studies of FH variant carriers. Thus, the discussion henceforth will focus on these loci, recognizing that additional mechanisms of monogenic hypercholesterolemia have been described [2]. Moreover, the evaluation and determination of FH variant pathogenicity is a lofty challenge in and of itself [10,11▪] and beyond the scope of this commentary.
Genotype versus phenotypeThe genomics era ushered in large-scale analyses of the clinical impacts of FH variants. Such studies clearly demonstrated an imperfect overlap between the population of FH variant carriers and the population with phenotypic FH. In one minimally selected healthcare system cohort that matched electronic health records (EHR) with exome-wide sequencing of 50 726 adults, 229 FH variant carriers were identified [12]. Although these individuals had higher LDL cholesterol (LDL-C) than noncarriers on average (+69 mg/dl in an age and sex adjusted model), there was substantial overlap in the observed distributions of maximum observed LDL-C in the EHR. Among 53 subjects who met Dutch Lipid Clinic Network criteria for Definite FH, 37 (70%) did not carry an FH variant. Conversely, among the 215 carriers of FH variant with sufficient data, only 51 (24%) met criteria for Definite or Probable FH. Further, 96 (44%) were classified as ‘Unlikely FH.’ This discrepancy between FH genotype and phenotype immediately raises the question of the relative importance of genotype versus phenotype with regards to atherosclerotic risk.
Variant risk and low-density lipoprotein cholesterolIn a landmark study, Khera et al.[13] examined LDL-C levels and FH variant carrier status across multiple coronary artery disease (CAD) case-control cohorts. They found carriers of FH variants were at increased risk for CAD across all LDL-C levels, including those with LDL-C < 130 mg/dl. Others subsequently corroborated that among cohorts with severe hypercholesterolemia, carriers of FH variants are at increased risk for atherosclerotic disease compared to noncarriers, even after adjusting for baseline LDL-C [14,15]. Similarly, among patients with clinically diagnosed FH, the subset who carry FH variants are at increased risk, independent of baseline LDL-C [16,17▪▪].
A fundamental limitation of these important studies is the use of a single LDL-C measurement in the risk models. FH is defined by high LDL-C from birth. Thus, differences in cumulative LDL-C between carriers and noncarriers may not be captured by a single LDL-C measurement in adulthood. Analysis of longitudinal data from the Atherosclerosis Risk in Communities study and the Framingham Heart Study Offspring cohort is consistent with this idea [13]. When comparing FH variant carriers to noncarriers matched on baseline LDL-C, age, sex, and statin use, carriers demonstrated higher longitudinal LDL-C compared to noncarriers. However, this analysis was limited by small sample size with only 25 FH variant carriers in total across the two cohorts.
This example highlights a major challenge in FH research. Prospective cohorts with significant longitudinal data are limited in size, making the number of FH variant carriers in these cohorts quite small. Mega biobanks now allow studies of FH variant carriers in much larger numbers, and the UK Biobank in particular has contributed to several novel and insightful studies [14,18–21,22▪▪]. However, such mega biobanks typically lack longitudinal lipid data. One important exception is the Million Veteran Program (MVP). MVP is a large diverse biobank that matches EHR data to genetic data [23]. Participants are United States veterans who receive medical care through Veterans Health Administration, a network of >1000 hospitals and clinics across the United States. This network shares a single EHR system that has been in continuous use for decades. Given these features, colleagues and I recently turned to MVP to examine FH variant risk and longitudinal LDL-C [24▪▪].
We identified 1504 FH variant carriers in a diverse cohort of 455 734 adults [24▪▪]. The mean age at enrollment was 62, and the median number of LDL-C measurements per subject was 12. Prescription data was used to determine which lipid measurements were taken in the setting of statin use, and untreated values were estimated by dividing treated values by 0.7 when indicated. Using the mean observed LDL-C for each subject provided better discrimination for identifying FH variant carriers [area under the curve (AUC) = 0.68] compared to using a single measurement (AUC = 0.64). Nonetheless, the observed distributions of mean LDL-C overlapped substantially between carriers and noncarriers, consistent with studies of single LDL-C measurements [12–14].
Examining the relationship between LDL-C exposure, FH variants, and risk for CAD required careful study design. The majority of CAD cases in MVP are prevalent (diagnosed prior to enrollment), and the time between a subject's earliest LDL-C measurement and date of diagnosis (or last follow-up) can vary. We therefore employed a nested case-control study design that used incidence density sampling to create case–control sets that were appropriately matched on etiologic exposure window [25]. Briefly, for each CAD case, we defined the exposure window as the time from first available LDL-C measurement to the time of first diagnosis of CAD. We then matched 10 controls per case such that the first available LDL-C for each control was measured at the same age as the case, and each control was at risk during the exposure window (i.e., had not accrued a CAD diagnosis). We additionally matched year of birth, sex, and ancestry group. Using logistic regression, we estimated the CAD risk associated with FH variants while adjusting for observed LDL-C over matched exposure windows. In total, we identified 23 091 cases matched to 230 910 controls. The average exposure window was 5.7 ± 4.6 years. FH variant carriers had 53% increased odds of CAD before adjusting for LDL-C [odds ratio (OR) 1.53, 95% confidence interval (CI) 1.24–1.89]. This risk was attenuated when adjusting for baseline (first) LDL-C (OR 1.41, 95% CI 1.14–1.74). The risk was even further attenuated with adjustment for the mean LDL-C over the exposure window (OR 1.33, 95% CI 1.07–1.64). Importantly, the study design conditions on survival to enrollment, leading to a survival bias. As such, the analysis underestimates the CAD risk associated with FH variants. However, the key result is the pattern rather magnitude of risk. We found that mean longitudinal LDL-C accounts for more FH variant risk than a single measurement, but independent genetic risk still remains. We observed this same pattern in both sex and ancestry stratified analyses and in several sensitivity analyses [24▪▪].
Others have examined LDL-C exposure in FH cohorts using the ‘cholesterol-year-score’ [26,27,28▪], which is calculated as follows:
LDL-C max × [age at diagnosis/initiation of statin] +
LDL-C at inclusion x [age at inclusion - age at diagnosis/initiation of statin therapy]
This simplistic measurement only considers two LDL-C values (untreated and treated) and assumes these levels are constant through time before and after initiation of treatment. In a recent small study of patients who were seen at a lipid clinic, FH causing LDLR variants were associated with an odds ratio for prevalent CAD of 2.44 (95% CI 1.31–4.56). After adjusting for cholesterol-year-score, the risk attenuated but remained significant (OR 2.14, 95% CI 1.12–4.07) [28▪].
FH variants in the general population confer risk for atherosclerotic cardiovascular disease beyond what is predicted by baseline or longitudinal LDL-C measurements during adulthood. This observation is important because it highlights the value of genetic testing as a means of better identifying high risk individuals [29]. Perhaps equally important, understanding why this pattern exists shines a light on the limitations of current clinical practice and lipid-based risk assessment.
Why variant risk is independent of clinically observed low-density lipoprotein cholesterolThe explanation for why the atherosclerotic risk associated with FH variants is independent of observed LDL-C is simple. LDL-C measurements in adults do not reflect lifetime exposure to LDL particles. In fact, even during adulthood, a single LDL-C measurement is only a moderately good proxy for adult longitudinal exposure. In a study of four prospective adult cohorts, index LDL-C had a Pearson correlation coefficient of 0.7 with either of two metrics of longitudinal LDL-C (cumulative LDL-C and time-weighted average LDL-C) [30▪▪]. In a multivariable Cox model adjusted for index LDL-C, both cumulative LDL-C and time-weighted average LDL-C remained independent risk predictors of incident cardiovascular disease [30▪▪]. Notably, metrics of LDL-C exposure may have even greater predictive value when measured earlier in life. A prospective study of young adults showed that cumulative LDLC in the third decade of life was more strongly associated with cardiovascular risk than that during the fourth decade [31]. The importance of cumulative LDL-C differences over time is also seen in clinical trial data. A meta-analysis of 21 trials of statins, ezetimibe, and PCSK9 inhibitors showed that the observed benefit of LDL-C reduction is not fixed but rather increases over the duration of LDL-C reduction [32▪▪]. The authors estimated that for 1 mmol/l (38.7 mg/dl) decrease in LDL-C, the relative risk reduction for major cardiovascular events after 1 year was 12% and increased steadily each year up to 29% at year 7. Given these analyses, it is easy to imagine that LDL-C during childhood and adolescence may have a profound impact on risk that is not completely reflected by LDL-C measurements in later life. Indeed, diagnosis and treatment of FH during childhood has been associated with a profound decrease in incident cardiovascular events compared to diagnosis/treatment later in life [33,34▪].
Considering an example helps to illustrate the issue of measured versus lifetime LDL-C. Figure 1 shows two hypothetical patients who have LDL-C of 140 mg/dl during routine clinical evaluation in their 40s. A risk model for atherosclerotic cardiovascular disease would attribute the same level of LDL-C risk to each of these two patients. However, patient 1 has had LDL-C of 140 mg/dl since birth, while patient 2 had average LDL-C during early life. LDL-C of 140 mg/dl is not considered severely elevated in adults, but such a level is >95–99th percentile during childhood [35,36]. The increased risk of patient 1 is not appreciated by observed LDL-C but would be captured by a marker of lifetime LDL-C exposure. No doubt, FH variants are markers of increased LDL-C exposure.
 FIGURE 1:
FIGURE 1: Illustration of how clinically observed LDL-C may not reflect lifetime cumulative LDL-C. In this hypothetical example, Patient 1 and patient 2 have the same LDL-C at routine assessments during middle age. Neither has measurements from childhood and adolescence, and differences in lifetime exposure are therefore not reflected in the clinical evaluation. Unobserved differences in cumulative LDL-C leads to risk that is not accounted for by observed LDL-C. LDL-C, low-density lipoprotein cholesterol.
This model explains the atherosclerotic risk associated with FH variant that is independent of observed LDL-C. In fact, any genetic change that impacts lifetime LDL-C should be expected to impact atherosclerotic risk beyond what can be appreciated by adult LDL-C measurements. The greater the effect on lifetime LDL-C, the greater the effect on risk independent of observed LDL-C. For example, when considering specific types of FH variants, loss-of-function variants of LDLR are associated with higher LDL-C compared to other FH variants. Likewise, such variants have been found to associate with greater atherosclerotic risk independent of baseline LDL-C compared to other variant types [37▪,38▪]. This greater magnitude of independent risk seen among carriers of LDLR null variants reflects a greater impact on lifetime LDL-C compared to other variant types. In the same vein, subjects who carry variants of uncertain significance (VUS) in FH loci have lower LDL-C compared to those who carry pathogenic variants but higher LDL-C compared to those who carry neither pathogenic variants nor VUS. Along with intermediary LDL-C, the VUS carriers also have intermediary risk compared to the other two groups [39]. Studies of common variants further corroborate this model. Among individuals with severe hypercholesterolemia who do not carry an FH variant, those who have a high polygenic score for LDL-C are at increased risk compared to those who do not have a high polygenic score, independent of baseline LDL-C [14]. Hence, polygenic risk for adult hypercholesterolemia impacts cumulative LDL-C exposure. At the other end of the LDL-C spectrum, genetic variants that lower LDL-C provide a greater protection against CAD than is expected based on the risk reduction per unit LDL-C reduction observed in statin trials [40]. These observations all result from the same model: genetic changes that impact lifetime cumulative LDL-C will influence atherosclerotic risk beyond what can be appreciated by adult LDL-C measurements.
PleiotropyThe discussion thus far has focused on how and why FH variants associate with atherosclerotic risk independent of observed LDL-C. However, it is worth considering an alternative possibility: pleiotropy. Pleiotropy exists when a gene plays an important role in more than one biological pathway. If FH loci are involved in pathways causal for atherosclerosis but unrelated to LDL-C, then we would expect to observe risk independent of LDL-C.
It has previously been hypothesized that FH variants may impact lipoprotein(a), given higher levels observed in clinical FH cohorts [41]. However, eloquent analysis using an unselected population cohort showed that the association between lipoprotein(a) and clinical FH is due to ascertainment bias, and FH variants do not cause elevated lipoprotein(a) [18]. This example demonstrates why observational studies of clinical FH populations can be misleading, and unbiased population studies are needed to determine whether FH variants are pleiotropic. In that setting, a powerful approach is the phenome-wide association study (PheWAS). PheWAS examines the relationship between a given exposure (e.g., FH variants) and the observed clinical outcomes in a population. A PheWAS of >200 000 MVP subjects of European ancestry tested the relationship between FH variants (748 carriers) and 1171 EHR-derived diagnoses. All significant associations were either related to dyslipidemia or CAD [42].
Similar results are easily generated using the publicly available GeneBass tool (https://app.genebass.org/) [43]. This tool allows users to broadly assess the impact of predicted loss-of-function variants at a given locus, considering >4500 phenotypes across nearly 400 000 subjects in the UK Biobank with whole-exome sequencing. Included phenotypes are extensive, encompassing medical records, questionnaires, and lab and imaging evaluations. Using this tool to assess LDLR loss of function variants results in several highly significant associations. The top hits in each category are all related to lipids and atherosclerosis (Fig. 2).
 FIGURE 2: PheWAS of LDLR predicted loss of function variants in the UK Biobank using the GeneBass online tools shows strong associations with lipid- and atherosclerosis-related phenotypes without evidence for pleiotropy. The LDLR gene was searched in GeneBass (https://app.genebass.org/), release date June 7, 2022. A standard burden test was used to assess predicted loss of function variants aggregated at the gene level. The tool considers 4529 phenotypes across 394 841 individuals with exome sequence data. Phenotypes are divided and colored by categories, with examples being lab measurements, diagnoses, surgeries, medication usage, etc. Top hits for LDLR all relate to lipids and atherosclerosis. For readability, only select associations are labeled. LDLR, low-density lipoprotein receptor gene.
FIGURE 2: PheWAS of LDLR predicted loss of function variants in the UK Biobank using the GeneBass online tools shows strong associations with lipid- and atherosclerosis-related phenotypes without evidence for pleiotropy. The LDLR gene was searched in GeneBass (https://app.genebass.org/), release date June 7, 2022. A standard burden test was used to assess predicted loss of function variants aggregated at the gene level. The tool considers 4529 phenotypes across 394 841 individuals with exome sequence data. Phenotypes are divided and colored by categories, with examples being lab measurements, diagnoses, surgeries, medication usage, etc. Top hits for LDLR all relate to lipids and atherosclerosis. For readability, only select associations are labeled. LDLR, low-density lipoprotein receptor gene.It is impossible to completely rule out pleiotropy for any given gene, and observational studies may occasionally report non-LDL associations with FH that could suggest pleiotropy. However, such observations must be carefully considered in the context of potential biases. Unbiased populations studies have so far provided no compelling evidence that FH variants have significant pleiotropic effects impacting atherosclerotic risk.
CONCLUSIONFH variants, by definition, impact LDL receptor mediated endocytosis of circulating LDL particles. This fundamental change in the physiology of LDL regulation leads to an increase in lifelong LDL-C compared to what would have been experienced in the absence of such a variant. It is through this increased exposure that FH variants cause increased risk for atherosclerotic cardiovascular disease. Adult measurements of LDL-C can only partially capture this increased cumulative exposure, and therefore these measurements only partially account for the risk associated with FH variants. Genetic testing provides valuable risk-stratifying information that cannot be replaced by standard clinical assessments. Moreover, these observations should motivate a greater focus on identifying and treating hypercholesterolemia earlier in life.
AcknowledgementsNone.
Financial support and sponsorshipNIH-CTSA grant KL2TR003143. S.L.C. is supported by NIH-CTSA grant KL2TR003143.
Conflicts of interestThere are no conflicts of interest.
REFERENCES AND RECOMMENDED READINGPapers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
REFERENCES 1. Brown MS, Goldstein JL. How LDL receptors influence cholesterol and atherosclerosis. Sci Am 1984; 251:58–69. 2. Berberich AJ, Hegele R A. The complex molecular genetics of familial hypercholesterolaemia. Nat Rev Cardiol 2019; 16:9–20. 3. Khera AV, Hegele RA. What is familial hypercholesterolemia, and why does it matter? Circulation 2020; 141:1760–1763. 4. Acta Med Scand, Müller C. Xanthomata, hypercholesterolemia, angina pectoris. Vol 95. 1938; 75–84. 5. Wilkinson CF, Hand EA, Fliegelman MT. Essential familial hypercholesterolemia. Ann Intern Med 1948; 29:671–686. 6. Stecher RM, Hersh AH. Note on the genetics of hypercholesterolemia. Science 1949; 109:61–62. 7. Innerarity TL, Weisgraber KH, Arnold KS, et al. Familial defective apolipoprotein B-100: low density lipoproteins with abnormal receptor binding. Proc Natl Acad Sci USA 1987; 84:6919–6923. 8. Varret M, Rabès JP, Saint-Jore B, et al. A third major locus for autosomal dominant hypercholesterolemia maps to 1p34.1-p32. Am J Hum Genet 1999; 64:1378–1387. 9. Abifadel M, Varret M, Rabès J-P, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet 2003; 34:154–156. 10. Iacocca MA, Chora JR, Carrié A, et al. ClinVar database of global familial hypercholesterolemia-associated DNA variants. Hum Mutat 2018; 39:1631–1640. 11▪. Chora JR, Iacocca MA, Tichý L, et al. The clinical genome resource (ClinGen) familial hypercholesterolemia variant curation expert panel consensus guidelines for LDLR variant classification. Genet Med 2022; 24:293–306. 12. Abul-Husn NS, Manickam K, Jones LK, et al. Genetic identification of familial hypercholesterolemia within a single U.S. health care system. Science 2016; 354:aaf7000. 13. Khera AV, Won H-H, Peloso GM, et al. Diagnostic yield and clinical utility of sequencing familial hypercholesterolemia genes in patients with severe hypercholesterolemia. J Am Coll Cardiol 2016; 67:2578–2589. 14. Trinder M, Francis GA, Brunham LR. Association of monogenic vs polygenic hypercholesterolemia with risk of atherosclerotic cardiovascular disease. JAMA Cardiol 2020; 5:390–399. 15. Tada H, Kawashiri M-A, Nohara A, et al. Impact of clinical signs and genetic diagnosis of familial hypercholesterolaemia on the prevalence of coronary artery disease in patients with severe hypercholesterolaemia. Eur Heart J 2017; 38:1573–1579. 16. Trinder M, Li X, DeCastro ML, et al. Risk of premature atherosclerotic disease in patients with monogenic versus polygenic familial hypercholesterolemia. J Am Coll Cardiol 2019; 74:512–522. 17▪▪. D’Erasmo L, Minicocci I, Di Costanzo A, et al. Clinical implications of monogenic versus polygenic hypercholesterolemia: long-term response to treatment, coronary atherosclerosis burden, and cardiovascular events. J Am Heart Assoc 2021; 10:e018932. 18. Trinder M, DeCastro ML, Azizi H, et al. Ascertainment bias in the association between elevated lipoprotein(a) and familial hypercholesterolemia. J Am Coll Cardiol 2020; 75:2682–2693. 19. Trinder M, Paquette M, Cermakova L, et al. Polygenic contribution to low-density lipoprotein cholesterol levels and cardiovascular risk in monogenic familial hypercholesterolemia. Circ Genom Precis Med 2020; 13:515–523. 20. Fahed AC, Wang M, Homburger JR, et al. Polygenic background modifies penetrance of monogenic variants for tier 1 genomic conditions. Nat Commun 2020; 11:3635. 21. Patel AP, Wang M, Fahed AC, et al. Association of rare pathogenic DNA variants for familial hypercholesterolemia, hereditary breast and ovarian cancer syndrome, and lynch syndrome with disease risk in adults according to family history. JAMA Netw Open 2020; 3:e203959. 22▪▪. Fahed AC, Wang M, Patel AP, et al. Association of the interaction between familial hypercholesterolemia variants and adherence to a healthy lifestyle with risk of coronary artery disease. JAMA Netw Open 2022; 5:e222687. 23. Gaziano JM, Concato J, Brophy M, et al. Million veteran program: a mega-biobank to study genetic influences on health and disease. J Clin Epidemiol 2016; 70:214–223. 24▪▪. Clarke SL, Tcheandjieu C, Hilliard AT, et al. Coronary artery disease risk of familial hypercholesterolemia genetic variants independent of clinically observed longitudinal cholesterol exposure. Circ Genom Precis Med 2022; 15:e003501. 25. Essebag V, Genest J, Suissa S, Pilote L. The nested case–control study in cardiology. Am Heart J 2003; 146:581–590. 26. Schmidt HH, Hill S, Makariou EV, et al. Relation of cholesterol-year score to severity of calcific atherosclerosis and tissue deposition in homozygous familial hypercholesterolemia. Am J Cardiol 1996; 77:575–580. 27. Gallo A, Charriere S, Vimont A, et al. SAFEHEART risk-equation and cholesterol-year-score are powerful predictors of cardiovascular events in French patients with familial hypercholesterolemia. Atherosclerosis 2020; 306:41–49. 28▪. Michikura M, Hori M, Ogura M, et al. The impact of gene variants on the thickness and softness of the Achilles tendon in familial hypercholesterolemia. Atherosclerosis 2022; 358:41–46. 29. Sturm AC, Knowles JW, Gidding SS, et al. Clinical genetic testing for familial hypercholesterolemia: JACC Scientific Expert Panel. J Am Coll Cardiol 2018; 72:662–680. 30▪▪. Zhang Y, Pletcher MJ, Vittinghoff E, et al. Association between cumulative low-density lipoprotein cholesterol exposure during young adulthood and middle age and risk of cardiovascular events. JAMA Cardiol 2021; 6:1406–1413. 31. Domanski MJ, Tian X, Wu CO, et al. Time course of LDL cholesterol exposure and cardiovascular disease event risk. J Am Coll Cardiol 2020; 76:1507–1516. 32▪▪. Wang N, Woodward M, Huffman MD, Rodgers A. Compounding benefits of cholesterol-lowering therapy for the reduction of major cardiovascular events: systematic review and meta-analysis. Circ Cardiovasc Qual Outcomes 2022; 15:e008552. 33. Luirink IK, Wiegman A, Kusters DM, et al. 20-Year follow-up of statins in children with familial hypercholesterolemia. N Engl J Med 2019; 381:1547–1556. 34▪. Tada H, Kojima N, Yamagami K, et al. Early diagnosis and treatments in childhood are associated with better prognosis in patients with familial hypercholesterolemia. Am J Prev Cardiol 2022; 12:100434. 35. Daniels SR, Greer FR. Committee on Nutrition. Lipid screening and cardiovascular health in childhood. Pediatrics 2008; 122:198–208. 36. Skinner AC, Steiner MJ, Chung AE, Perrin EM. Cholesterol curves to identify population norms by age and sex in healthy weight children. Clin Pediatr (Phila) 2012; 51:233–237. 37▪. Tada H, Kojima N, Yamagami K, et al. Effects of different types of pathogenic variants on phenotypes of familial hypercholesterolemia. Front Genet 2022; 13:872056. 38▪. Paquette M, Carrié A, Bernard S, et al. Effect of the LDL receptor mutation type on incident major adverse cardiovascular events in familial hypercholesterolaemia. Eur J Prev Cardiol 2022; 29:2125–2131. 39. Tada H, Kojima N, Yamagami K, et al. Impact of variants of uncertain significance of LDL receptor on phenotypes of familial hypercholesterolemia. J Clin Lipidol 2022; 16:863–869. 40. Ference BA, Yoo W, Alesh I, et al. Effect of long-term exposure to lower low-density lipoprotein cholesterol beginning early in life on the risk of coronary heart disease: a Mendelian randomization analysis. J Am Coll Cardiol 2012; 60:2631–2639. 41. Alonso R, Andres E, Mata N, et al. Lipoprotein(a) levels in familial hypercholesterolemia: an important predictor of cardiovascular disease independent of the type of LDL receptor mutation. J Am Coll Cardiol 2014; 63:1982–1989. 42. Sun YV, Damrauer SM, Hui Q, et al. Effects of genetic variants associated with familial hypercholesterolemia on low-density lipoprotein-cholesterol levels and cardiovascular outcomes in the million veteran program. Circ Genom Precis Med 2018; 11:e002192. 43. Karczewski KJ, Solomonson M, Chao KR, et al. Systematic single-variant and gene-based association testing of thousands of phenotypes in 394 841 UK Biobank exomes. Cell Genomics 2022; 2:100168.
留言 (0)