
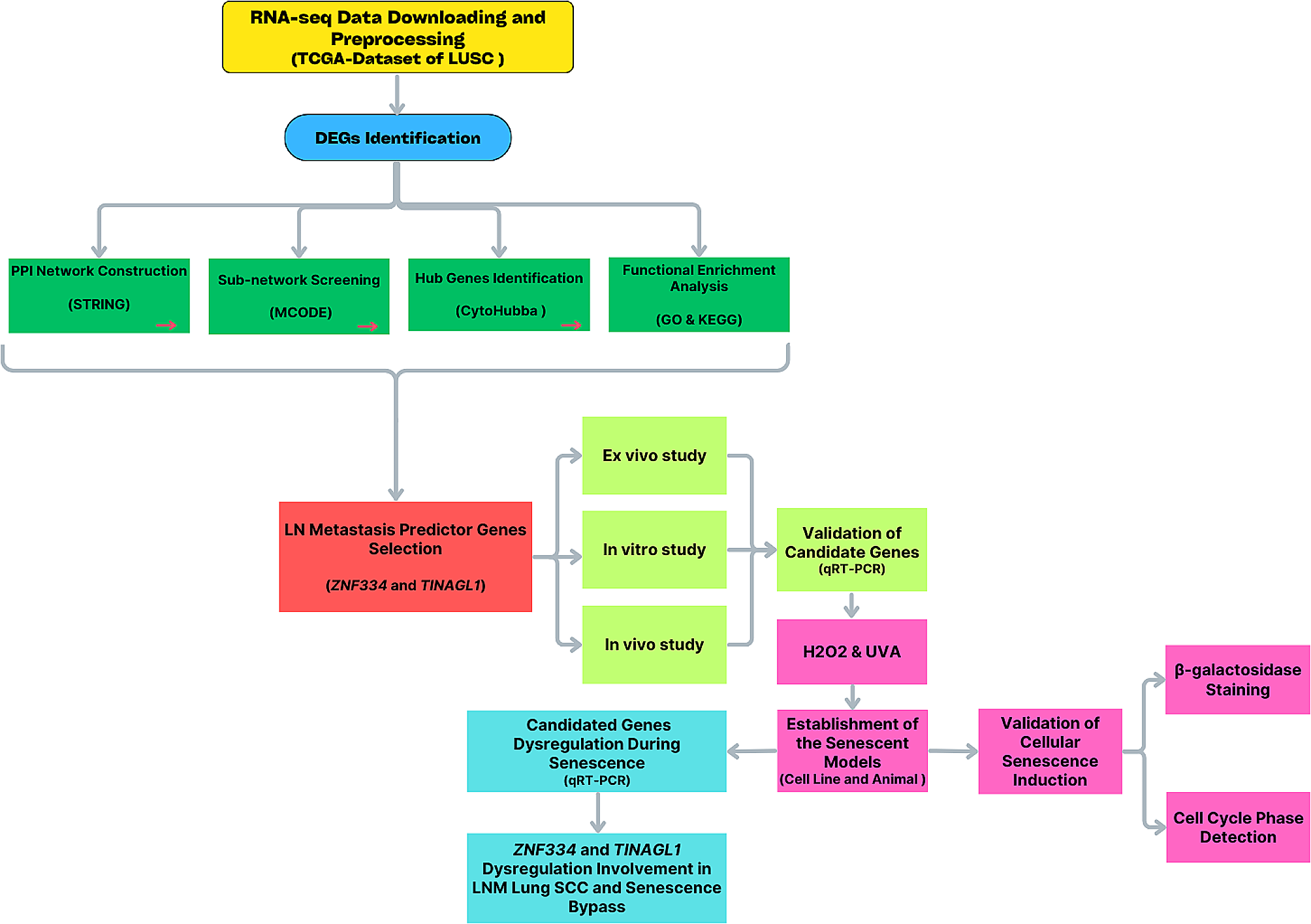
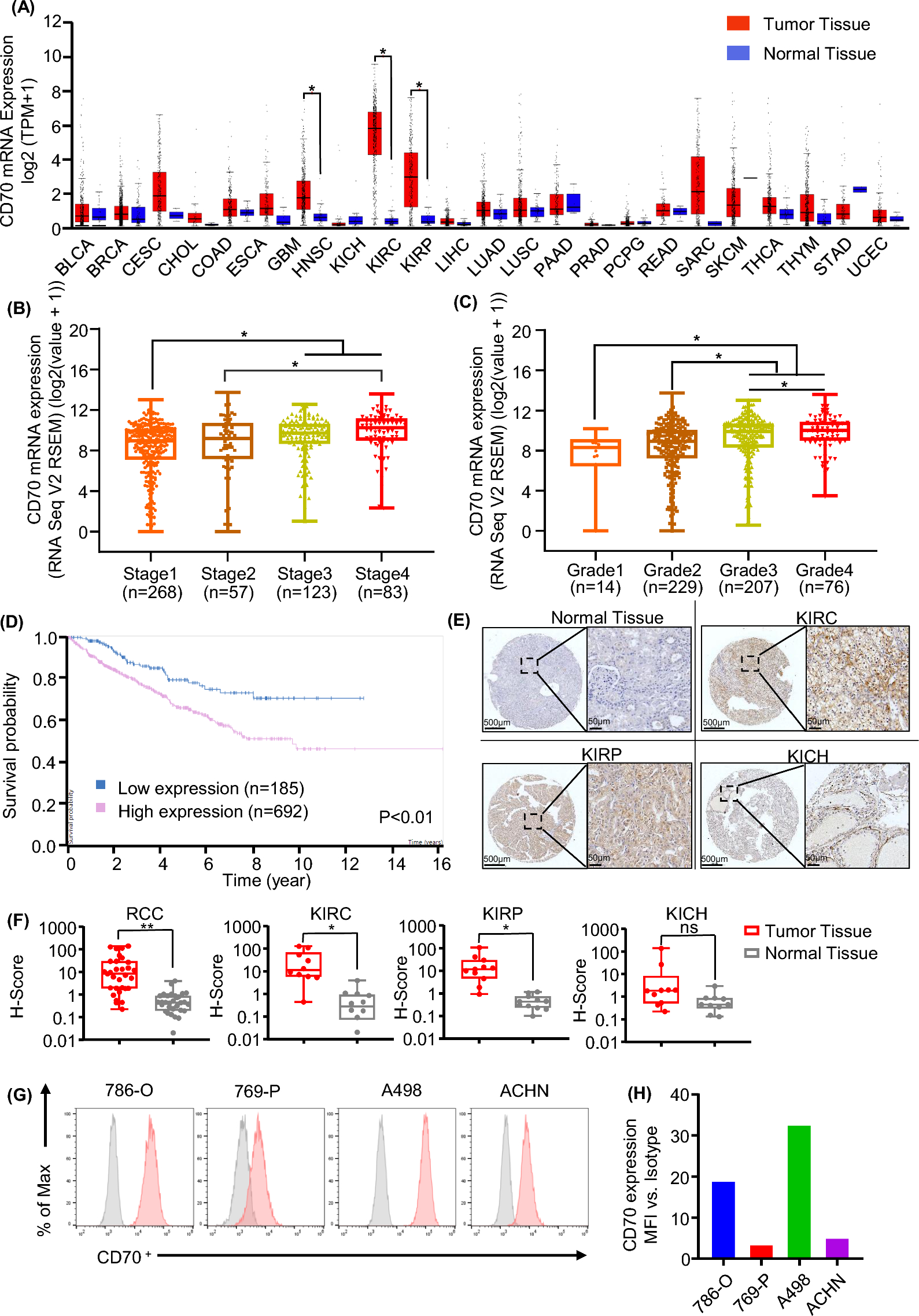
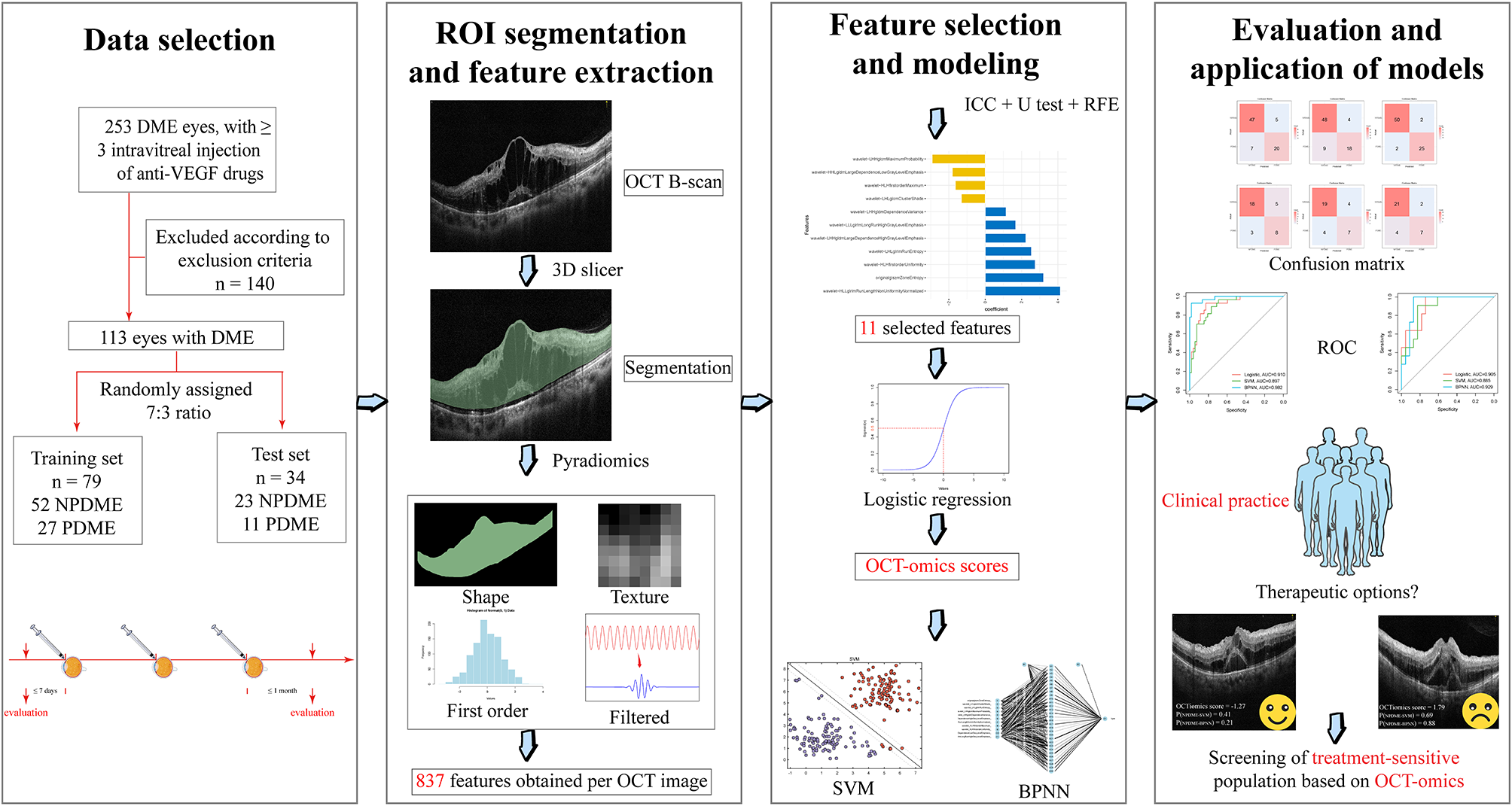
記住我


Following a rigorous pathological diagnosis and exclusion process, 72 fecal samples from 34 HCC-cirrhosis patients, 20 cirrhosis patients, and 18 healthy controls were collected and analyzed using ITS2 rDNA sequencing. Benefiting from a uniform sample collection protocol, all stool samples were yellow and soft. The clinical characteristics of the participants, such as age, gender, and body mass index (BMI), were matched among the three groups (Table 1). The patients with HCC and cirrhosis also have similar Child-Pugh and cirrhotic etiological compositions. Serum levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), total bilirubin (TBIL), and direct bilirubin (DBIL) were significantly elevated, whereas albumin and platelet levels were significantly reduced in HCC patients compared with healthy controls (Table 1). Compared to the cirrhosis group, the albumin level was significantly lower in the HCC group, while the remaining serum parameters were not significantly different (Table 1).
Table 1 Clinical characteristics of the enrolled participantsAltered community composition in HCC patients compared to healthy controls and hepatocirrhosis patientsFirst, we assessed the composition of the gut fungi in our cohort. The rank abundance curves revealed that the species have good richness and uniformity in each group (Fig. 1A). The Venn diagram showed that 1961 OUTs were shared among the three groups. 2698 OTUs, 2655 OTUs, and 1084 OTUs were unique to the HCC group, cirrhosis group, and healthy controls, respectively, implying that HCC patients have the greatest abundance of unique OTUs (Fig. 1B). The fungal alpha diversity, as estimated by the Shannon diversity and Simpson diversity, was significantly reduced in patients with cirrhosis compared to healthy controls (P = 0.04 and 0.04 respectively; Fig. 1C, D). The above indicators also displayed a decreasing tendency in fungal diversity from the healthy controls to the HCC patients, but the difference was not significant (P = 0.08 and 0.14, respectively; Fig. 1C, D). However, there was no significant difference in diversity between the two patient cohorts (P = 0.65 and 0.25, respectively; Fig. 1C, D).
Fig. 1
Diversity analysis in the HCC, LC, and HC groups. A Rank abundance curves. B Venn diagram displaying the overlap of OTUs identified among the three groups. Alpha diversity was estimated by the Shannon index C and Simpson index D. The distributional difference of gut mycobiota profiles was assessed using PCoA E and NMDS F based on a weighted_unifrac matrix. HCC hepatocellular carcinoma, LC liver cirrhosis, HC healthy controls
The beta diversity was next applied to explore the fungal compositions among the three groups by performing weighted_unifrac matrix. The PCoA showed that the three groups of individuals formed a fairly good separation of gut fungi (Fig. 1E). In addition, NMDS is applied to visualize the distances among the three groups, as shown in Fig. 1F, illustrating the distinct separation. MRPP analysis further confirmed the above findings (HCC vs. cirrhosis: P = 0.048; HCC vs. control: P = 0.041; cirrhosis vs. control: P = 0.021). Taken together, these results indicate the presence of altered intestinal fungal composition in patients with HCC.
Gut fungal dysbiosis in HCC-cirrhosis, including increased levels of C. albicans and Malassezia sp.As shown in Fig. 2A, B, the cluster heatmap showed the differentially enriched fungal microbiotas among the three groups at the genus and species levels. Next, the Wilcoxon rank-sum test was used to explore differences at the genus and species levels. The results showed that there were 32 and 47 significantly differential taxa at the genus and species levels between HCC patients and healthy controls (Additional file 4: Table S3). The eight most abundant of the above differential taxonomic units were presented in Fig. 2C, D. We found that the relative abundances of Candida, C. albicans, Malassezia, Malassezia sp., Rhizopus, Neocatenulostroma, Neocatenulostroma sp., and F. proliferatum were significantly higher in HCC patients compared to healthy controls, while the relative abundances of Actinomucor, A. elegans, Mucor, M. circinelloides, Alternaria, A. alternata, Trichocladium, and P. mandshurica were significantly lower (Fig. 2C, D). We further analyzed the fungal differences between HCC patients and cirrhosis patients, finding a significant enrichment of Candida, C. albicans, C. tropicalis, Monographella, M. nivalis, Bipolaris, Bipolaris sp., Nakaseomyces, Nakaseomyces sp., Malassezia, Malassezia sp., Sporothrix, S. ramosissima, Staphylotrichum, S. coccosporum and depletion of Archaeorhizomyces in the HCC group (Fig. 2E, F, Additional file 5: Table S4). These data point to the presence of fungal dysbiosis in HCC patients, which might be linked to hepatocellular carcinogenesis.
Fig. 2
Differential analysis of fungal communities in the HCC, LC, and HC groups. A Heatmap of fungal abundance clustering at the genus level A and species level B. The eight most abundant of the differential taxonomic units at the genus level C and species level D between HCC patients and healthy controls. The eight most abundant of the differential taxonomic units at the genus level E and species level F between HCC patients and cirrhosis patients. The Wilcoxon rank-sum test was used. HCC hepatocellular carcinoma, LC liver cirrhosis, HC healthy controls
To identify the specific fungal taxa associated with HCC, we further compared the fungal composition between the HCC patients and healthy controls using LEfSe. As shown in Fig. 3A and Additional file 8: Figure S1A, the intestinal mycobiota of HCC patients were enriched in Saccharomycetales fam Incertae sedis, Candida, C. albicans, Malasseziomycetes, Malasseziales, Malasseziaceae, Malassezia, and Malassezia sp.; while the gut mycobiome of healthy controls was elevated in Pleosporaceae, Alternaria, A. alternata, Mucoromycota, Mucoromycetes, Mucorales, Mucoraceae, Actinomucor, A. elegans, Mucor, and M. circinelloides. The distinct taxa at the species level were displayed as relative abundance histograms (Fig. 3B–E). Besides, we also found that there were significant differences in the fungal abundance between the HCC patients and cirrhosis-only patients. The fifteen taxa, including Saccharomycetes, Saccharomycetales, Nakaseomyces, Nakaseomyces sp., Saccharomycetales fam Incertae sedis, Candida, C. albicans, Xylariales, Malasseziomycetes, Malasseziales, Malasseziaceae, Malassezia, Malassezia sp., Bipolaris, and Bipolaris sp. were increased in the HCC group (Fig. 3F–N), while Archaeorhizomycetes, Archaeorhizomycetales, Archaeorhizomycetaceae, Archaeorhizomyces, Archaeorhizomyces sp., Archaeorhizomycetes sp., Hyponectriaceae, Monographella, M. nivalis, Lysurus, and L. cruciatus were highly enriched in the cirrhosis group (Fig. 3F–N). These differentially abundant taxa can be considered potential biomarkers.
Fig. 3
The differential taxa in the HCC, LC, and HC groups using the linear discriminant analysis effect size (LEfSe) analysis. A LDA scores were computed for differentially abundant taxa in the gut fungi of HCC patients and healthy controls. F LDA scores were computed for differentially abundant taxa in the gut fungi of HCC patients and cirrhosis patients. Length indicates the effect size associated with a taxon. P = 0.05 for the Kruskal-Wallis sum-rank test; LDA score > 4. B–E, G–N The histogram of the relative abundance distribution of each taxon at the species level. HC healthy controls, HCC hepatocellular carcinoma, LC liver cirrhosis, LDA linear discriminant analysis
Correlation between gut mycobiota with TNM stage and clinical parameters in HCC patientsTo further elucidate the relationship between fungal disorders and tumor progression, we used the LEfSe method to estimate differences in fungal taxa between 19 HCC patients with TNM stage III-IV and 15 HCC patients with TNM stage I-II (8th edition of the AJCC/UICC TNM staging system). The clinical characteristics of HCC patients with different TNM stages are detailed in Additional file 6: Table S5. HCC Patients with TNM stage III-IV demonstrated a significantly higher relative abundance of Saccharomycetales fam Incertae sedis, Candida, C. albicans, Archaeorhizomycetes, Archaeorhizomycetales, Archaeorhizomycetaceae, Archaeorhizomyces, Archaeorhizomyces sp. than the HCC patients with TNM stage I-II. Moreover, Saccharomycetaceae, Nakaseomyces, Nakaseomyces sp., Saccharomyces, S. cerevisiae were significantly more abundant in HCC patients with TNM stage I-II (Fig. 4A, B).
To understand the relationship between the intestinal fungi and clinical physiological parameters in HCC patients, Spearman analysis was implemented. The top 20 taxa with the highest relative abundance at the genus level were selected for analysis. Data showed that nutritional indicators and liver damage indicators significantly correlated with some fungi (Fig. 4C). For example, the abundance of Candida was significantly positively related to the level of TBIL and negatively associated with red blood cell (RBC) and hemoglobin, indicating that Candida is associated with impaired liver function and poorer nutritional status. Further, CCA analysis of the clinical parameters screened according to VIF and BioENV showed that albumin and gamma-glutamyltransferase (GGT) were important drivers of fungal distribution in HCC patients (Fig. 4D, Additional file 7: Table S6).
Fig. 4
Correlation of gut mycobiota with TNM stage and clinical physiological indicators in HCC patients. A Taxonomic cladogram from LEfSe showing differences in fecal taxa of HCC patients with stage III-IV and stage I-II. B LDA scores were computed for differentially abundant taxa in the gut fungi of HCC patients with stage III-IV and stage I-II. Length indicates the effect size associated with a taxon. P = 0.05 for the Kruskal-Wallis sum-rank test; LDA score > 4; C Spearman correlation analysis of fungal taxa and clinical physiological indicators in HCC patients. D Canonical correspondence analysis (CCA) of clinical indicators correlated with the fungal profile of patients with HCC. HCC hepatocellular carcinoma, LDA linear discriminant analysis, ALT alanine aminotransferase, AST aspartate aminotransferase, ALP alkaline phosphatase, GGT gamma-glutamyltransferase, TBIL total bilirubin, DBIL direct bilirubin, TBA total bile acid, RBC red blood cell, Hb hemoglobin, PA prealbumin, TP total protein, ALB albumin, GLB globulin, WBC white blood cell, NeuC neutrophil count, LYMC lymphocyte count, MonoC monocytes count, LMR monocyte-to-lymphocyte ratio, NLR neutrophil-lymphocyte ratio, PLT platelets, PT prothrombin, PT-act prothrombin time activity, PT-INR the international normalized ratio of prothrombin, APTT activated partial thromboplastin time, TT thrombin time, AT-III antithrombin III, *P < 0.05, **P < 0.01
Most of the patients included in this study had a history of HBV infection, so we further analyzed the intestinal fungal differences between 27 patients with HCC and 16 patients with cirrhosis, and those patients had a history of HBV infection. As shown in Additional file 9: Figure S2, the gut mycobiota of HCC patients was enriched in Candida, C. albicans, Bipolaris, Bipolaris sp., Nakaseomyces, Nakaseomyces sp., Sporothrix, S. ramosissima, Malassezia, Malassezia sp., etc., while the gut mycobiota of cirrhosis was elevated in Monographella, M. nivalis, Lysurus, L. cruciatus, Thermoascus, T. aurantiacus, etc.
Classification of the HCC group compared to the non-HCC groupWe further explored the diagnostic ability of the top eight fungal microbials that showed the most significant differences among three groups (Fig. 2D, E). The ROC curves displayed diagnostic potential for some of these fungi, including Candida albicans (AUC = 0.749, Fig. 5A), Malassezia sp. (AUC = 0.789, Fig. 5B), Neocatenulostroma sp. (AUC = 0.711, Fig. 5C), Nakaseomyces sp. (AUC = 0.676, Fig. 5D), Candida tropicalis (AUC = 0.655, Fig. 5E), Alternaria alternata (AUC = 0.655, Fig. 5F), and Pichia membranifaciens (AUC = 0.639, Fig. 5G). The combined models (Candida albicans, Malassezia sp. and Neocatenulostroma sp.) confirmed the powerful discriminatory ability of intestinal flora in distinguishing HCC patients from healthy controls and cirrhosis (AUC = 0.906, Fig. 5H). However, the above findings are yet to be confirmed by future multicenter studies with large sample sizes.
Fig. 5
Classification of the HCC group compared to the non-HCC group. ROC curves of Candida albicans A, Malassezia sp. B, Neocatenulostroma sp. C, Nakaseomyces sp. D, Candida tropicalis E, Alternaria alternata F, and Pichia membranifaciens G. H ROC curves analysis to evaluate the classification ability of the gut mycobiome signature (combined Candida albicans, Malassezia sp., and Neocatenulostroma sp.) in predicting different groups. ROC receiver operating characteristic, AUC area under the curve
Functional classification prediction of the specific taxonomicBecause of lacking a powerful tool for annotating the function of fungi, we concentrated on the functional guilds of the fungal microorganisms instead, using FUNGuild. As shown in Fig. 6A, B, the clustered heatmap revealed distinctively differential functions among HCC patients, cirrhotic patients, and healthy controls. Specifically, the pathotrophs, such as plant pathogen, animal pathogen-undefined saprotroph, and animal pathogen-endophyte-plant saprotroph-soil saprotroph, were significantly enriched in the HCC group (Fig. 6A, B); the saprotrophs, including soil saprotroph and dung saprotroph-soil saprotroph, and animal pathogen-endophyte-plant pathogen-wood saprotroph were remarkably increased in the cirrhosis group (Fig. 6B); while the saprotrophs, such as soil saprotroph and undefined saprotroph-wood saprotroph, and ectomycorrhiza were significantly enriched in the healthy controls (Fig. 6A). Thus, our analysis indicates that the symbiotic ecological relationships of gut fungi are altered in patients with HCC and are dominated by pathological parasitism, which can receive nutrients from and adversely affect host cells.
Fig. 6
Functional classification predictions. Fungal functional annotations between the HCC and healthy controls A and between the HCC and liver cirrhosis B were performed by FUNGuild. Fungi were divided into different categories at the Guild levels according to the ways of absorption and utilization of environmental resources. HCC hepatocellular carcinoma, LC liver cirrhosis, HC healthy controls
C. albicans and M. furfur promote the progression of HCCTo confirm whether C. albicans and M. furfur are involved in HCC development, we treated C57BL/6 mice with oral gavage of PBS, C. albicans, or M. furfur and inoculated them with subcutaneous tumors (Fig. 7A, B). We found that after 2 weeks post exposure, the tumor volume and weight were significantly increased in the C. albicans and M. furfur groups, while there was no significant difference in the body weight among the three groups (Fig. 7C–E, Additional file 10: Figure S3). IHC staining confirmed that more proliferative cells were observed in the xenografts of the C. albicans and M. furfur groups as indicated by Ki-67 staining (Fig. 7F, G). These results indicate that abnormal colonization by C. albicans and M. furfur contributes to HCC development.
Fig. 7
Abnormal colonization of C. albicans and M. furfur can promote HCC development. A Schematic diagram of the oral gavage intervention protocol using phosphate-buffered saline, C. albicans or M. furfur for C57BL/6 mice (6 mice per group). The images of tumor-bearing mice B and tumor masses C. Tumor volume D and weight E were compared among the three groups at the end of the experiment. F Representative images of subcutaneous tumor among the three groups immunostained with Ki67 (200 ×). G Quantification of Ki67 + cells. *P < 0.05, **P < 0.01, Wilcoxon rank sum test was used
留言 (0)