
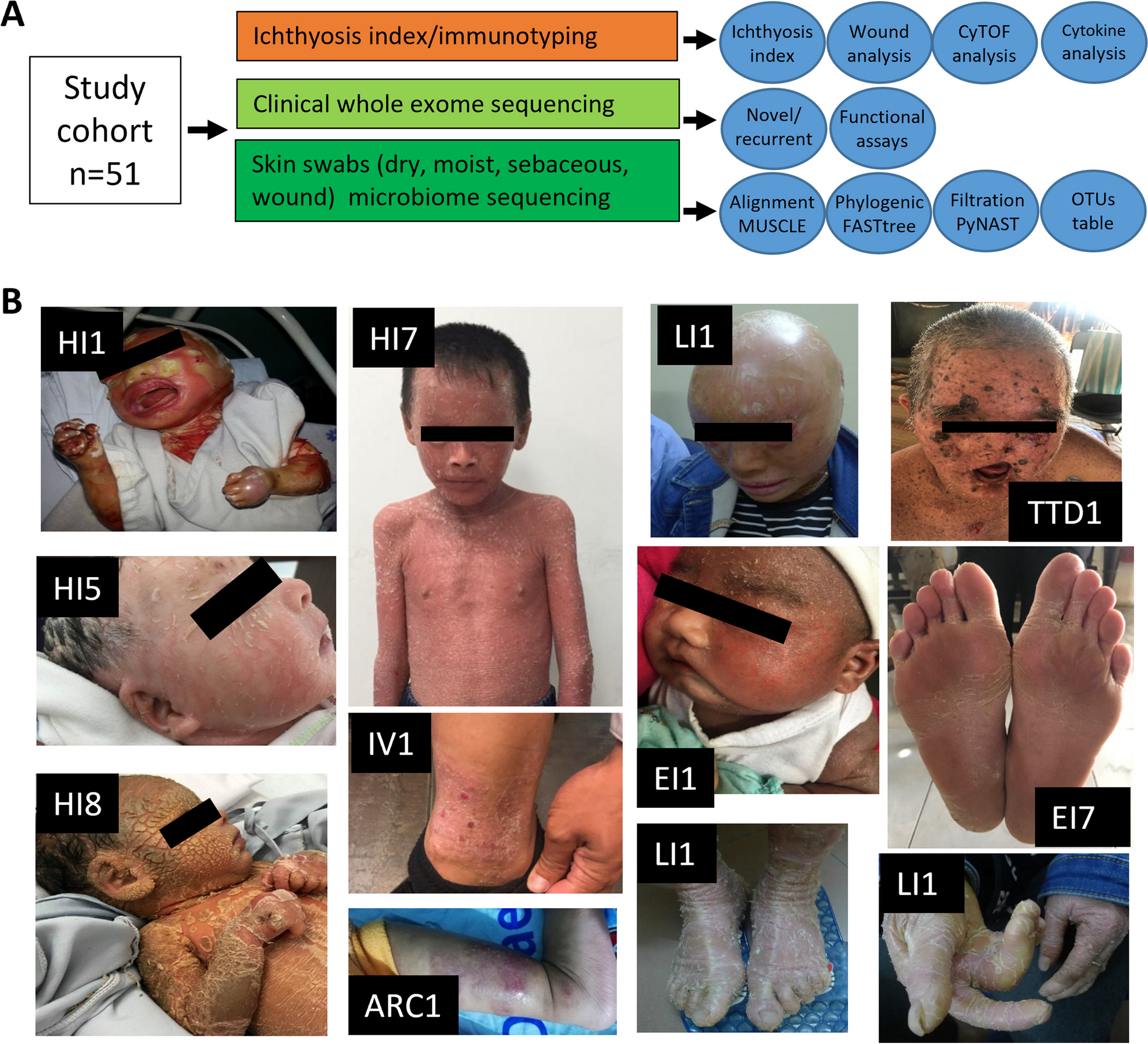
記住我
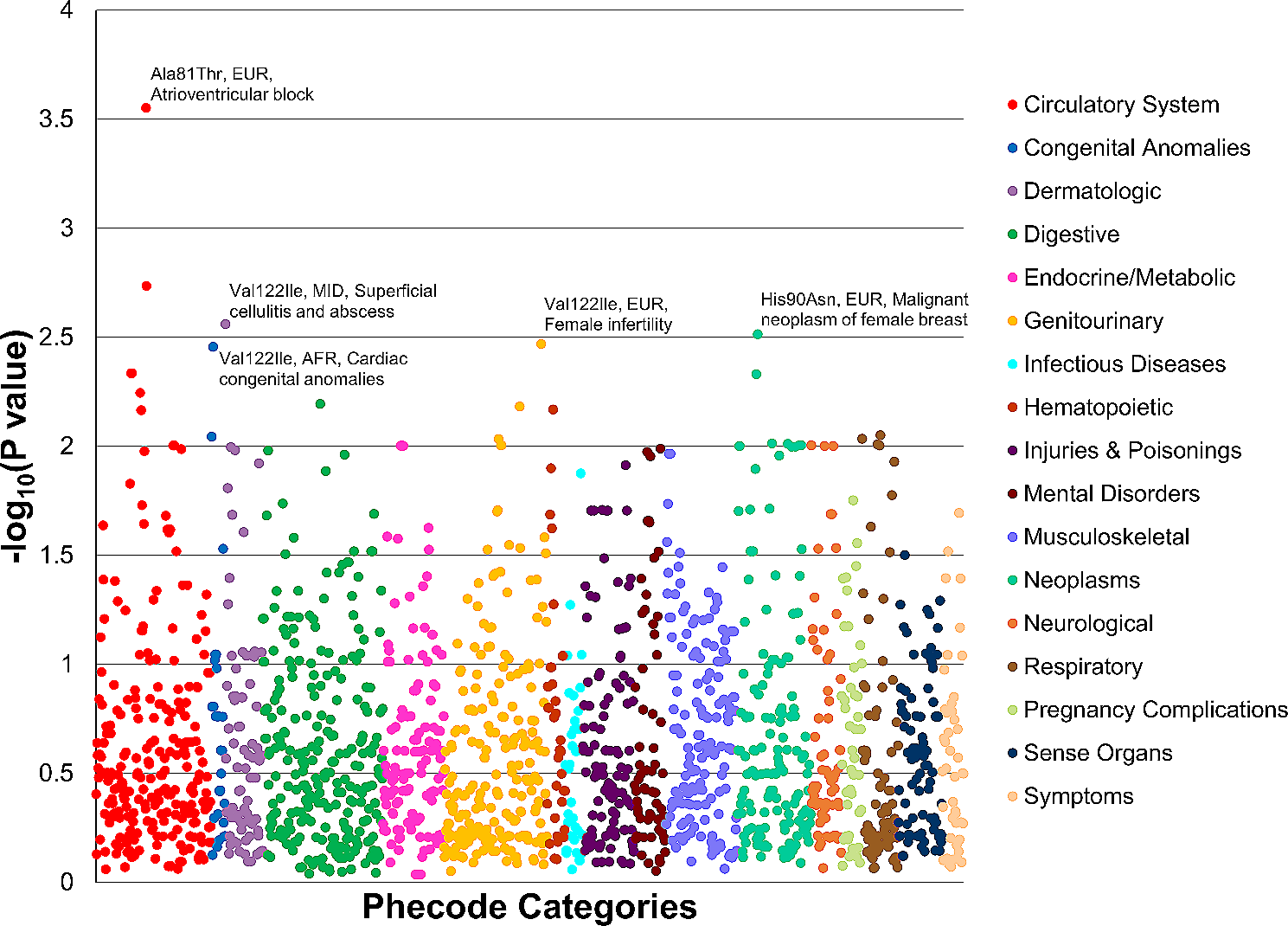
We developed SpliceAI-visual, which displays SpliceAI’s RS on a genome browser. SpliceAI-visual betterments compared to SpliceAI are summarized in Table 1.
Table 1 SpliceAI-visual solves some SpliceAI limitationsOvercoming the DS pitfallAs already stated, the value of 0.2 is recommended by the authors of SpliceAI as a threshold for the four DS to discriminate potential splice-altering variants from non-altering variants. We present several examples demonstrating the relevance of SpliceAI-visual when the DS are low.
Examples from the literatureIdentifying pseudo-exon inclusion SCN1AThe deep intronic substitution NM_001165963.4(SCN1A):c.4002 + 2461 T > C (Table 2, Fig. 1) has been demonstrated by minigene assays to induce the exonization of an out-of-frame 64-bp intronic sequence [17]. This 64-bp exonization mechanism has not been elucidated, but was correctly identified by SpliceAI with low DS (AG: 0.18; DG: 0.15). Using SpliceAI-visual, we show that while the DS are below the recommended threshold, the RS for the wild-type sequence are already significant (acceptor site: 0.64; donor site: 0.73). This results in high RS for the variant sequence T > C (acceptor site: 0.82; donor site: 0.87) and finally in the inclusion of the intronic sequence in the transcript. The mRNA proportion aberrant/normal transcript was not estimated.
Table 2 HGVS descriptions, SpliceAI, SpliceAI-visual scores, and ACMG classification of the variants analyzed in this study MFGE8Similarly, the pathogenic variant NM_005928.4(MFGE8):c.871-803A > G is responsible for the inclusion of an intronic sequence containing a stop codon (Table 2, Fig. 2) [18]. Again, the SpliceAI DS are low (AG: 0.15; DG: 0.16), but the reference allele was already identified with mild RS.
Fig. 2
The delta score (DS) pitfall: discrepancy between SpliceAI’s DS and SpliceAI raw scores (RS). SpliceAI-visual outputs of MFGE8 deep intronic variant displayed in IGV. Above: SpliceAI RS for the reference allele of MFGE8; below: SpliceAI RS for the pathogenic variant NM_005928.4(MFGE8):c.871-803A > G functionally attested to cause the exonization of an intronic sequence containing a stop codon (red). Orange: acceptor site prediction; Blue: donor site prediction. The variant position is pointed by a dashed line
SpliceAI-visual identified the resulting acceptor and donor sites on the variant allele as strong candidates (respectively, 0.84 and 0.74), and the use of a graphical output (bedGraph files) loaded in a genome browser allowed a quick identification of the termination codon using the three frames translation track in IGV or in the UCSC Genome Browser. This intronic inclusion was estimated to be ~ 10 times more abundant than the wild-type transcript.
Unpublished cases SETD5: enhancing the retention of a “poison” exonGenome-trio sequencing of patient 1 revealed a de novo variant in intron 17 of SETD5: NM_001080517.3:c.2476 + 198A > C (Table 2, Fig. 3). SpliceAI DS were low with an AG and DG of 0.05 and 0.04, respectively. However, those DS were added to high RS (acceptor: 0.94, donor: 0.95) as shown by SpliceAI-visual. Indeed, we observed a low level of intronic retention in RNAseq of controls. This intronic retention of 97 bp led to the inclusion of a premature stop codon and a presumed degradation by NMD. By performing RNAseq from a blood sample of the patient, we showed that the intronic retention of this “poison” exon was dramatically enhanced compared to 2 controls. The variant was found in 95% of the reads, confirming the causal effect of our variant on this retention.
Fig. 3
The delta score pitfall: SETD5 poison exon retention caused by an intronic substitution. RNAseq and SpliceAI-visual outputs displayed in IGV. Above: SpliceAI RS for the reference allele of SETD5, along with one control individual; below: SpliceAI RS for the pathogenic deep intronic variant NM_001080517.3:c.2476 + 198A > C, along with RNAseq of patient 1. Orange: acceptor site prediction; Blue: donor site prediction. The variant position is pointed out with a dashed line. Although the variant A > C is heterozygous, 95% of RNAseq reads carry the C, suggesting the causative role of this allele in the retention
GRN: guiding functional investigationsSpliceAI-visual is also convenient for guiding functional investigations. The following heterozygous variant NM_002087.4(GRN):c.-9A > G (Table 2) was identified in a 70-year-old male with Fronto-Temporal Dementia (patient 2), and plasmatic progranulin values compatible with a monoallelic alteration of GRN (see Sup Methods and Patients). This variant was previously identified in another affected patient, but the authors failed to evidence any abnormal splicing products [19]. This variant is predicted by SpliceAI to weaken the canonical donor site of this first 5’UTR exon (donor loss of 0.48). The initial RT-PCR has been performed on fibroblasts, but the exonic primers (F1-R1) failed to identify any abnormal products, as previously reported, even in the presence of an NMD inhibitor. Thanks to SpliceAI-visual, we were able to spot the putative rescuing donor site, which was predicted with a modest gain of + 0.19, but added to an RS of 0.75 on the reference allele (Fig. 4). This prediction was in favor of a 271-bp intronic retention. Another reverse primer (R2) has been designed in the predicted intronic 271-bp retention and showed amplification in the patient, and not in control individuals. The failure of the initial exonic RT-PCR (F1-R1) to amplify both wild-type and retention fragments could be due to the competitive advantage of the short fragment over the fragment including the 271-bp retention.
Fig. 4
The delta score pitfall: extending the 5’UTR of GRN. RNAseq and SpliceAI-visual outputs displayed in IGV. Above: SpliceAI RS for the reference allele of GRN along with RNAseq from one control; below: SpliceAI RS for NM_002087.4(GRN):c.-9A > G, along with RNAseq of patient 2. Bottom: two upstream Open Reading Frames in the intronic retention (yellow), height corresponding to the initiation strength of the AUG codon based on the Kozak context from TIS [20]
Adjusting the PVS1 criteriaAccording to the standard guidelines of the American College of Medical Genetics and Genomics (ACMG), the PVS1 criteria includes “canonical +/− 1 or 2 splice sites in a gene where the loss of function is a known mechanism of disease” [21]. However, alteration of a canonical splice site can result in other non-truncating consequences by various mechanisms: (1) an in-frame exon skipping (initially stated in the caveats of the aforementioned guideline), (2) an in-frame deletion by the creation of an exonic rescuing splice site, or (3) an in-frame intronic retention devoid of in-frame stop codon [22, 23]. We show here with various cases the relevance of SpliceAI-visual in the assessment of the PVS1 criteria relative to variants altering canonical splice sites.
CASKWe report the case of a 9-year-old male individual, presenting with learning disabilities and microcephaly (see Additional file 1: Methods and Patients, patient 3). Solo-exome sequencing showed a hemizygous substitution in a canonical donor site of the gene CASK, NM_003688.3(CASK):c.172 + 1G > A, absent from control databases (gnomAD, deCAF) [24, 25]. No other pathogenic or likely pathogenic variant was retained. This donor site disruption affects the MANE transcript of CASK. This hemizygous variant of patient 3 is predicted by SpliceAI to result in a DL, along with a + 0.71 DG. With SpliceAI-visual, this DG was predicted to lead to in-frame retention of 18 bp (6 amino acids, no stop codon, Fig. 5). Furthermore, this donor’s DS of + 0.71 adds to a probability of 0.28 on the reference allele, resulting in an RS of 0.99 on this donor site (Fig. 5). In accordance with SpliceAI-visual predictions, RT-PCR on peripheral blood of patient 3 identified the 18-bp retention on 100% of transcripts (Fig. 5), which precluded the use of the Very_Strong weight of the PVS1 criteria. Without the very strong weight, this variant couldn’t be classified as likely pathogenic or pathogenic. The significance of this variant was classified as Uncertain (Table 2).
Fig. 5
Scaling down the PVS1 criteria of a canonical splice site variant in CASK. Segregation, RT-PCR and SpliceAI RS of NM_003688.3(CASK):c.172 + 1G > A, hemizygous in patient 3. This variant leads to the complete in-frame retention of 18 bp (no wild-type 297 bp product was observed in patient 3 RT-PCR lane), as predicted by SpliceAI-visual. This 18-bp retention does not include stop codon and is predicted to insert 6 amino acids
KMT2DThe variants NM_003482.4(KMT2D):c.5189-1G > C and c.5782 + 1G > A (Table 2) are located in canonical splice sites of KMT2D and solely on this argument, the PVS1 criteria could apply, as loss-of-function variants are a known mechanism of KMT2D-related Kabuki syndrome. Based on this argument, these variants have recently been submitted as Likely Pathogenic in ClinVar (VCV001496460.1, VCV001506261.1) [26]. Surprisingly, these variants were reported in unaffected individuals in the general population (c.5189-1G > C is absent from gnomAD v2.1.1 / v3.1.2, but found in 11 individuals in UK Biobank exomes [24, 27]. c.5782 + 1G > A is present in 3 heterozygous individuals in gnomAD v2 and v3) [24], which is inconsistent with the penetrance and severity of monoallelic KMT2D loss-of-function variants (OMIM: 147,920). This discrepancy could be explained by splicing rescue, which was well predicted by SpliceAI-visual (Fig. 6).
For c.5189-1G > C, SpliceAI-visual shows the creation of an in-frame rescuing acceptor site, predicted to delete 8 poorly conserved residues.
For c.5782 + 1G > A, SpliceAI-visual predicts the complete loss of the donor site (− 1), and a modest gain of an in-frame nearby donor site (+ 0.28). This modest gain is another example of the DS pitfall (see above), adding on to a cryptic site predicted with an RS of 0.71 on the reference allele, resulting in an RS of 0.99 on the alternate allele. Moreover, this donor-rescuing site results theoretically in the inclusion of 3 amino acids in the final product, which may have less deleterious consequences and explain the presence of this variant in gnomAD.
Fig. 6
Scaling down the PVS1 criteria of canonical splice site variants in KMT2D. Left: another a priori PVS1 variant NM_003482.4(KMT2D):c.5189-1G > C, present in 11 individuals in UK Biobank. This variant is predicted to result in an in-frame rescuing acceptor site, deleting 8 poorly conserved amino acids. Right: SpliceAI-visual outputs and BAM from one heterozygous from gnomAD of NM_003482.4(KMT2D):c.5782 + 1G > A. This variant is present in 3 individuals in gnomAD, which is not consistent with the penetrance of loss-of-function variants of KMT2D. Also, the mild rescuing DS of 0.28 is added to a nonzero RS on the reference allele (delta score pitfall) and is predicted to result in a complete rescue of this donor site, with the in-frame retention of 9 bp
For c.5189-1G > C, SpliceAI-visual shows the creation of an in-frame rescuing acceptor site, predicted to delete 8 poorly conserved residues.
For c.5782 + 1G > A, SpliceAI-visual predicts the complete loss of the donor site (− 1), and a modest gain of an in-frame nearby donor site (+ 0.28). This modest gain is another example of the DS pitfall (see above), adding on to a cryptic site predicted with an RS of 0.71 on the reference allele, resulting in an RS of 0.99 on the alternate allele. Moreover, this donor-rescuing site results theoretically in the inclusion of 3 amino acids in the final product, which may have less deleterious consequences and explain the presence of this variant in gnomAD.
TTNWe describe here a similar case occurring in the TTN gene. NGS analyses targeted on congenital myopathy and muscular dystrophy gene panels identified in patient 4 (see Suppl. Methods for the phenotypic description) a variant in intron 116 of TTN: NM_001267550: c.31439-1G > C (Table 2) absent in the general population (gnomAD, deCAF) [24, 25] and predicted to affect splicing in exon 117. This variant located in the exon/intron junction of exon 117 is predicted to completely abolish the natural acceptor site, whereas the graphical output of SpliceAI-visual clearly shows a cryptic acceptor site located 9-bp downstream of the natural site (Fig. 7). Its use would lead to a 9-bp in-frame loss in exon 117, which has been confirmed by the RNAseq experiments (77 reads supporting the cryptic junction out of 222 reads (34.6%). Interestingly, SpliceAI-visual reveals a non-total raw probability of 0.53 to this rescuing acceptor site. Moreover, SpliceAI predicts the reduced strength of the natural donor site, located on the other side of exon 117. Taken together, these elements suggest a partial skipping of exon 117, which is further supported experimentally, as the exon 116–118 junction is attested by one read on RNAseq, and not seen in the two controls (Fig. 7). In the absence of a parental segregation study (no parents available) for dominant hypothesis, and of a second identified variant for recessive hypothesis, and regarding the RNAseq results, this variant was classified as a variant of uncertain significance (class 3).
Fig. 7
Scaling down the PVS1 criteria of a canonical splice site variant in TTN. RNAseq and SpliceAI-visual outputs displayed in IGV showing the predicted exon skipping (top view), and the in-frame rescue (bottom view). Top tracks: SpliceAI RS for the reference allele of TTN along with RNAseq from 2 controls; bottom tracks: SpliceAI RS for the NM_001267550.2(TTN):c.31349-1G > C along with RNAseq of patient 4
SETD5The following variant in SETD5 was identified in patient 5 in the heterozygous state, NM_001080517.3(SETD5):c.568-31_568dup p.(Asn190IlefsTer20) (Table 2), inherited from his asymptomatic mother. This 31-bp duplication is absent from gnomAD or deCAF [24, 25]; it duplicates the exon–intron border of exon 8 of SETD5 and is considered to have a high truncating impact according to SNPEff and VEP annotators [28, 29]. Indeed, this variant duplicates the acceptor site, resulting in two competing nearby acceptor sites: the first being out-of-frame—hence the predicted frameshift—and the second being in-frame. SpliceAI-visual, however, shows the second site to be the strongest, predicting no splicing alteration (Fig. 8), which was confirmed by RNAseq.
Fig. 8
Scaling down the PVS1 criteria of a putative frameshift in SETD5. SpliceAI-visual outputs displayed in IGV showing the predicted benign splicing outcome of this putative frameshift
Interpreting complex delinsFinally, SpliceAI-visual allows the interpretation of complex variants. For example, the following variant is a complex deletion–insertion variant occurring on an exon–intron border in the gene NM_001142800.2(EYS):c.2992_2992 + 6delinsTG (Table 2). However, most SpliceAI current public implementations or pre-computed whole genome VCFs currently do not process complex delins variations (i.e., other than deletion, insertion, or substitution), nor does Pangolin. Of note, those complex variations are handled by CI-SpliceAI but with numerical results [12]. The functional study of this variant by a minigene assay has shown the skipping of an entire out-of-frame exon [30]. We show that this exon skipping is well predicted by SpliceAI-visual (Fig. 9). In addition, we have tested SpliceAI-visual’s ability to predict 13 other complex delins, all of which were functionally attested to alter splicing, and correctly predicted by SpliceAI-visual (Additional file 1: Table S1).
Fig. 9
SpliceAI-visual outputs displayed in IGV showing the predicted exon skipping resulting from the complex delins NM_001142800.2(EYS):c.2992_2992 + 6delinsTG. Top track: SpliceAI RS for the reference allele of EYS; bottom track: SpliceAI RS for the delins in EYS
留言 (0)