
記住我
To identify patterns of age-related change in a data-driven manner, we developed a novel computational approach which we refer to here as Data-driven Identification and Classification of Nonlinear Aging Patterns (DICNAP). Figure 1 shows a graphical abstract depicting our methodology. In addition to age information for the subjects, input data consist of the methylome dataset, which is a matrix with subjects by DNA methylation site, with the elements of the matrix representing methylation intensities. Briefly, the workflow consists of the following steps.
The first step involves performing a nonlinear correlation analysis to identify which of the methylation sites in the genome-wide DNA methylation site are associated with age. We used the maximal information coefficient (MIC), which is a nonlinear correlation index that ranges from zero to one, to capture any type of association between two variables [14]. We calculated the MIC between age and methylation intensity for each site, as well as the associated P value using permutation tests. Sites with Benjamini–Hochberg adjusted P values < 0.05 were considered to be age-associated sites for the downstream analysis [15]. All other sites were considered to be non-correlated sites (NC).
The second step is to classify the identified age-associated sites into linear and nonlinear patterns. MIC - \(\rho ^2\) can be used as an index of nonlinearity, where \(\rho\) is the Pearson correlation coefficient [14]. A larger MIC - \(\rho ^2\) represents higher nonlinearity. We consider sites with a nonlinear index < 0.05 to be sites with a linear pattern. Among these linear sites, those with Pearson correlation coefficients \(\ge\) 0 were considered to be linearly increasing (LI), and sites with Pearson correlation coefficients < 0 were considered to be linearly decreasing (LD). All other age-associated sites were defined as having a nonlinear (NL) pattern.
The third step is a nonparametric function estimation for the nonlinear age-associated methylation sites. To focus on the difference in the shape of the functions, we applied standard normalization of the methylation intensity for each site. We estimated the function for age-related changes by nonparametric regression with spline smoothing. As a result, the functional data for each site were generated as a scaled intensity on a grid separated by one year of age.
The fourth step is a functional principal component analysis (FPCA), which is used to embed the nonlinear age-associated methylation sites into a low-dimensional space based on the function patterns. FPCA takes a set of functional data as an input and gives principal component (PC) scores for each function. The eigenfunction shows the principal pattern that each PC represents. In this step, FPCA is applied to the age-related functions after the previous step and embeds the sites into a PC coordinate space. The PCs with a contribution rate > 1% were adopted as the top PCs for downstream analysis.
The final step comprises a multivariate analysis using PC coordinates for NL sites. Once the methylation sites are embedded into the PC coordinate space, ordinary multivariate analysis can be applied. Clustering of the PC coordinates allows for data-driven classification of DNA methylation sites with nonlinear patterns. We used K-means clustering and classified nonlinear sites into groups. These groups were named NL1, NL2...NLK, where K is the number of clusters automatically detected. For each of the K clusters, the median of the PC coordinate values was set to the center, and the typical pattern for each cluster was reproduced based on the PC coordinate values of the center, mean function and eigenfunctions.
Finally, our workflow classifies all methylation sites into NC, LI, LD, NL1, NL2...NLK, and the representative function pattern for NL sites. The details of the classification method are described in the Methods section. We applied the proposed method to a simulation dataset and describe the results of the simulation data analysis in Additional File 1: Section 1. Briefly, the simulation study showed that DICNAP can appropriately identify potential functional patterns and classify sites from an aging methylome dataset. However, a limitation of the method is that nonlinear changes that begin in very old age are difficult to detect.
Fig. 1
The outline of DICNAP method. Input data are a methylome dataset consisting of a matrix with subjects by DNA methylation site, and the elements of the matrix represent methylation intensities. Age information for the subjects is also required. After the multistep procedure, all methylation sites are classified as NC, LI, LD, NL1, NL2...NLK
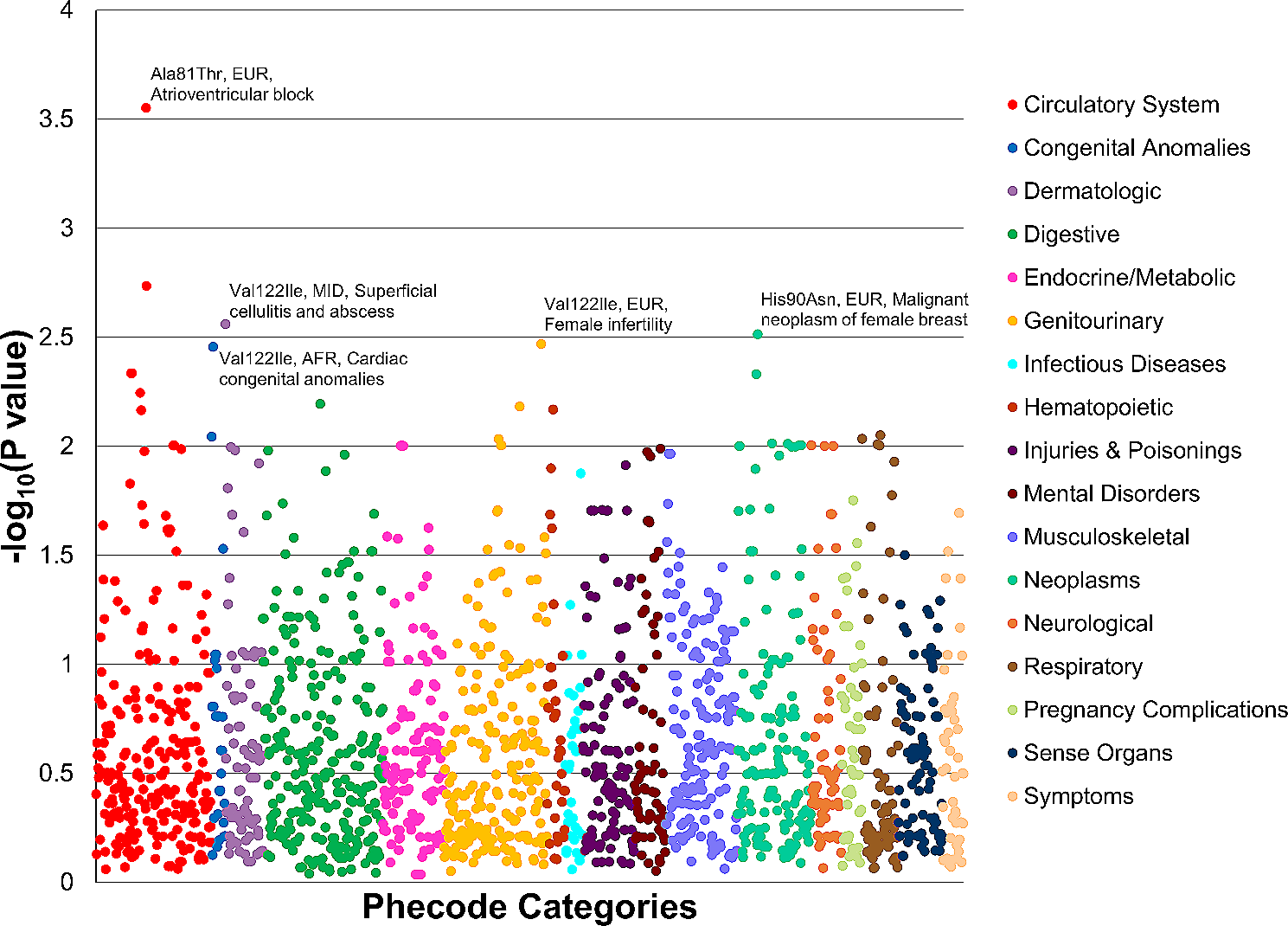
Genome-wide landscape of linear and nonlinear patterns for age-related changeWe obtained a large-scale DNA methylome dataset of human blood from two previous aging studies from the NCBI Gene Expression Omnibus (GEO) database (GSE87571 and GSE40279) [12, 13]. The male and female datasets were analyzed separately because previous studies have reported that age-related changes in DNA methylation differ between men and women [16]. To eliminate SNPs and other effects, multimodal sites were excluded from the downstream analysis. Finally, from the GSE87571 dataset, the methylation intensity beta values for 479,461 sites from 388 female subjects between the ages of 14 and 94 were used to compile Dataset1F, and those for 479,634 sites from 341 male subjects between the ages of 15 and 87 were used to compile Dataset1M for downstream analysis. Similarly, from the GSE40279 dataset, the methylation intensity beta values for 454,171 sites from 338 female subjects between the ages of 21 and 101 were used to compile Dataset2F, and those for 454,852 sites from 318 male subjects between the ages of 19 and 96 were used to compile Dataset2M. Details of the dataset preprocessing steps are described in the Methods section. Among these four datasets, we used Dataset1F as the main dataset in our analysis, because it has the largest sample size (n=388). We used the three other datasets to evaluate the stability of the analysis. We applied DICNAP to all four datasets; the results for the main dataset (Dataset1F) are described in Fig. 2, and those for the other three datasets are described in Additional File 2.
Figure 2a shows a plot of the MICs and Pearson correlation coefficients. Sites with a high MIC tended to be plotted with a bias toward regions with high Pearson’s correlation coefficient, which suggests that the changes in the DNA methylation intensity during aging were dominated by monotonous changes. This finding, which is consistent with the high performance of previously reported biomarkers for aging based on linear relationships, was also found for the three other datasets.
Figure 2b shows a Quantile–Quantile plot (QQ plot) of the nonlinear index defined by \(MIC - \rho ^2\). A null distribution was generated by subject label permutation. In this QQ plot, both inflation and deflation of the nonlinear index are observed. The inflation of the QQ plot shows that there are some methylation sites with nonlinear associations. The deflation of the QQ plot shows the existence of methylation sites with stronger linearity. These trends were also commonly observed in the other three datasets (Additional File 2), suggesting that some methylation sites have nonlinear monotonous intensity changes in addition to the linear changes. The breakdown of association types (NC, LI, LD, NL) in Dataset1F is shown in Fig. 2c. The percentage of age-associated sites (LI, LD, NL) was 27.7%, while those of Dataset1M, Dataset2F, and Dataset2M were 31.8%, 11.1%, and 4.1%, respectively. Although the same workflow and thresholds were applied to all of the datasets, differences in statistical power were observed across datasets.
To investigate biological phenomena associated with methylation sites that showed a linear response to age, we subjected LI and LD sites to Gene Ontology (GO) analysis. Additional File 3 shows all of the significant GO terms (false discovery rate (FDR) < 0.05). A total of 204 GO terms are significantly represented in the LI group, with the top five GO terms being “central nervous system neuron differentiation,” “cell fate commitment,” “DNA-binding transcription activator activity, RNA polymerase II-specific,” “pattern specification process,” and “synaptic membrane.” These GO terms were also significantly enriched in Dataset1M . In Dataset2F and Dataset2M, no GO terms were significantly enriched in the LI groups. In the LD group, no GO terms showed significant enrichment in any of the four datasets . The results for the other three datasets are shown in Additional File 4.
Figure 2d shows a bar plot of the contribution rate for PC1-PC5 in the main dataset. Most of the differences in function are explained by PC1 and PC2. Additional File 5 shows the mean function and eigenfunction for the FPCA for all four datasets. Except for the sign difference, their shapes are similar, although the mean function and \(\psi _1(Age)\) behave differently, mainly at the edges. Figure 2e shows the results for the PC coordinate values with the identified group labels in the main dataset; four groups (NL1, NL2, NL3, and NL4) were identified as having nonlinear patterns. Indeed, in all datasets, the number of groups with nonlinear-type sites was four. Figure 2f shows the representative functions for the NL groups. While the representative functions were somewhat unstable among the four datasets, age-related hypomethylation patterns that moderated after around age 60 (NL1 and NL4 in Dataset1F) appeared as nonlinear patterns in all of the datasets (NL1 and NL3 in Dataset1M, NL2 in Dataset2F, and NL1 in Dataset2M).
We investigated the stability of the classification of NL sites by calculating the absolute Pearson correlation coefficients for the PC1, PC2, and PC3 values for the methylation sites classified as NL in all four datasets (Additional File 6). PC1 is more strongly correlated in the four datasets (Pearson’s correlation coefficient 0.94 - 0.98) than PC2 and PC3. Since the clustering of the NL group is explained mainly by PC1 (Fig. 2e), the classification of the methylation sites for NL patterns is considered to be stable across datasets.
We applied GO analysis to nonlinear site groups (NL1, NL2, NL3, and NL4). From the representative functions, NL1 and NL4 have nonlinearly decreasing patterns. In the NL1 group, the top five GO terms were “passive transmembrane transporter activity,” “regulation of membrane potential,” “extracellular matrix,” “transporter complex,” and “regulation of trans-synaptic signaling.” In the NL4 group, the top five GO terms were “regulation of cell morphogenesis,” “regulation of ion transmembrane transport,” “cell-cell junction,” “extracellular matrix structural constituent,” and “transmembrane receptor protein kinase activity.” All of these top GO terms were significantly enriched in one of the nonlinear decreasing patterns (NL1 or NL3) in Dataset1M. In contrast, in Dataset2F, only a total of five GO terms were significant for the nonlinear decreasing groups (either N1 or NL2) and contained no top GO terms in the main dataset. In Dataset2M, no GO terms were significant for the nonlinear decreasing groups (either N1 or NL2). The full results of the GO analysis using the main and other datasets are presented in Additional File 3 and Additional File 4, respectively.
In the main dataset, NL2 and NL3 are characterized by having nonlinearly increasing patterns. In NL2, the top five GO terms are “central nervous system neuron differentiation”, “cell fate commitment”, “appendage development”, “morphogenesis of a branching structure”, and “regulation of animal organ morphogenesis”. In NL3, the top five GO terms are “embryonic organ development,” “pattern specification process,” “cell fate commitment,” “synapse organization,” and “mesenchyme development.” These top GO terms were all also included in NL1 or NL3 of Dataset1M and are characterized by having increasing patterns. GO terms other than presynapse were also included in NL3 of Dataset2F and NL4 of Dataset2M, which are groups characterized by a nonlinear increasing pattern in each analysis.
Fig. 2
Analysis of methylation intensity function by age in Dataset1F. a Plots of MIC (X-axis) and Pearson’s correlation coefficient (Y-axis). Each dot represents a methylation site. b QQ plot of nonlinearity index. The nonlinearity index was defined as \(MIC- \rho ^2\). The null distribution was generated by label permutation. The red line represents y = x. c Pie chart showing the genome-wide methylation sites for age-related association types: NC (non-correlated), LI (linear increase), LD (linear decrease), NL (nonlinear) . d Contribution rate for PC1–PC5. e PC1 and PC2 for 1000 randomly selected sites. The colors represent groups detected by clustering. f Representative functions for the identified NL groups (X-axis: age, Y-axis: scaled intensity)
We checked which groups contained known DNA methylation sites as biomarkers for aging in previous studies. For example, cg16867657 (annotated to ELOVL2), cg06639320 (annotated to FHL2), and cg16419235 (annotated to PENK) are known biomarkers for aging [17]. These sites showed positive linear correlations with age in the original study. In our analysis, these three sites were classified as LI in all four datasets.
In another study, cg02228185 (annotated to ASPA), cg25809905 (annotated to ITGA2B), and cg17861230 (annotated to PDE4C) were all shown to predict age by a linear model [18]. cg02228185 is grouped in LD in Dataset1F, Dataset1M, and Dataset2F. In Dataset2F, it is classified as NL1, which is a nonlinearly decreasing pattern. cg17861230 is grouped in LI in Dataset1F and Dataset1M, while it is grouped as NC in Dataset2F and NL4 in Dataset2M. cg25809905 is grouped in NL4 in Dataset1F; while it is LD in Dataset1M, NL1 in Dataset2F and NL1 in Dataset2M. In all three sites, the direction of increase and decrease is consistent with the original study. However, these sites did not always show linear associations, and in some cases, a nonlinear association was suggested.
We investigated which group the sites consisting Horvath’s epigenetic clock have been classified. Genome-wide DNA methylation-based biomarkers of aging are called epigenetic clocks. It has been demonstrated that the predicted age obtained using a linear model of the genome-wide methylation markers can predict chronological age [19]. In particular, Horvath’s epigenetic clock is a linear predictive equation consisting of 353 CpG sites and works with methylome data from a variety of human organs, suggesting a universal mechanism for why we age that transcends organ differences [20]. We therefore checked the associations among our identified groups and Horvath clock sites.
Figure 3 shows a breakdown of association types (NC, LI, LD and NL) in the Horvath clock sites in Dataset1F. The percentage of age-associated sites among the Horvath clock sites was higher than that in the genome-wide methylation sites. While Horvath clock sites included a higher percentage of age-associated sites, they also included a meaningful percentage of NC and NL as well as LI and LD. This suggests that methylation sites in the Horvath clock include nonlinear changes and sites with only small or no stand-alone associations. The results for the other three datasets support this (Additional File 7).
Fig. 3
Pie charts for age-related correlation patterns of Horvath clock sites in Dataset1F. The left panel is a pie chart of all sites and is the same as in Fig. 2c. The right panel is a pie chart of the sites included in the Horvath clock. NC: non-correlated, LI: linear increase, LD: linear decrease, NL: nonlinear
Applying the workflow to variability functionsGiven that aging causes loss of control over biomolecular expression, the variability in biomarker expression should change during aging. When the variability in the DNA methylation intensity changes during aging, the residuals of the fitted regression function are expected to reflect this change. Previous studies have shown age-related increases in the variability of DNA methylation [10, 21,22,23]; however, it is not clear how this variability changes with age. We, therefore, applied the DICNAP to analyze the pattern of variability functions for methylation intensity.
The DICNAP-based variability function analysis was applied as follows. The analysis was performed for age-associated sites (LI, LD, and NL). We applied nonparametric regression for all age-associated sites, computed the squares of the residuals, and created a squared residual matrix (Fig. 4a). The model for nonparametric regression is the same as with the intensity function analysis. We applied the DICNAP method to this matrix and classified the age-associated sites as varNC, varLI, varNL1, varNL2...varNLK in terms of variability. We conducted a simulation analysis and found that this approach effectively identified and classified the variability functions (Additional File 1: Section 2). In addition, the results also showed that linear variability changes tend to be identified as nonlinear, but these representative patterns can be identified as linear in downstream FPCA analyses.
Figure 4 shows the results of the variability function analysis for age-related sites in Dataset1F. Figure 4b shows a plot of the MICs and Pearson correlation coefficients. The plots are asymmetric and tend to have more sites with a positive correlation between variability and age. Figure 4c shows a QQ plot of the nonlinear index, suggesting the existence of nonlinear variability changes. This pattern was also identified in the other three datasets (Additional File 8).
After the classification of association types, no varLI and varLD sites were detected, and 15.6% of age-associated sites in Dataset1F had varNL characteristics (Fig. 4d). In Dataset1M, Dataset2F, and Dataset2M, 11.3%, 10.9%, and 15.4% of age-associated sites showed age-related variability changes, respectively. These findings suggested that about 10 \(\sim\) 15% of age-associated sites stably show age-associated variability changes.
Subsequent FPCA analysis identified patterns of variability function in the methylome dataset. The shape of the mean function and eigenfunction was similar among all four datasets (Additional File 9). Figure 4e shows a bar plot of the contribution rate of principal coordinates in the main dataset. Figure 4f shows the results for PC coordinate values with clusters identified in the main dataset. Three groups (varNL1, varNL2, and varNL3) were identified as having a nonlinear variability pattern. Figure 4g shows the representative functions for the NL groups. As representatives, we identified patterns (varNL1 and varNL3) in which the variability increases monotonically and accelerates after middle age. This pattern was also identified in the other three datasets.
Fig. 4
Analysis of variability function for age in Dataset1F. a Outline of the variability analysis using DICNAP. b Plots of MIC (X-axis) and Pearson’s correlation coefficient (Y-axis). Each dot represents a methylation site. c QQ plot of nonlinearity index, which is defined as \(MIC- \rho ^2\). The null distribution was generated by label permutations. The red line represents y = x. d Pie charts for the genome-wide methylation sites of age-related correlation patterns: NC (non-correlated), LI (linear increase), LD (linear decrease), NL (nonlinear). e Contribution rate for PC1–PC5. f PC1 and PC2 in the FPCA analysis for 1000 randomly picked NL sites. The colors represent the groups detected by clustering. g Representative age-related functions for nonlinear sites (X-axis: age, Y-axis: scaled variability)
留言 (0)