
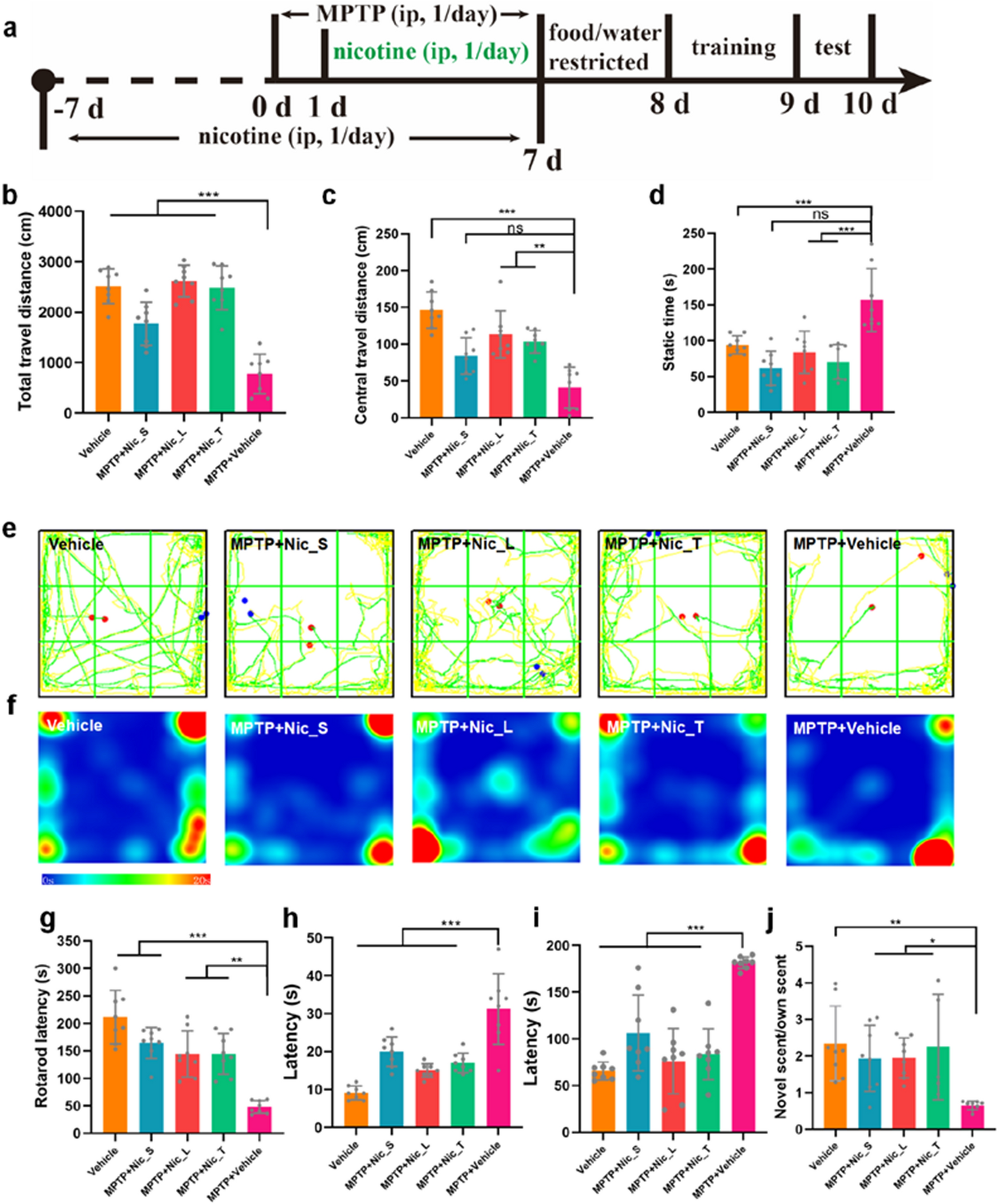
記住我
To construct the dynamic APA landscape in glomeruli between DN patients and control subjects, we utilized two different algorithms, DaPars [33] and QAPA [34], to identify APA events directly using RNA-seq datasets of glomeruli isolated from 50 biopsy-proven DN patients and 25 controls (Fig. 1a). PDUI and DPAU values were respectively calculated using DaPars and QAPA methods to measure the proportion of distal PAS usage for each gene in DN and control samples (Additional file 4: Table S3.1 and S3.2; Additional file 2: Fig. S2a). PCA analysis based on PDUI (Fig. 1b, top) and DPAU (Fig. 1b, bottom) values showed that the significant difference in 3′UTR lengths clearly distinguished DN glomeruli from the control, indicating the existence of dynamic APA events in DN and control glomeruli. Then, the ΔPDUI and ΔDPAU scores between DN and control were obtained to detect DN-associated 3′UTR length alterations (Additional file 4: Table S3.1 and S3.2). As shown in Fig. 1c and d (top), DaPars revealed 2835 genes with 3′UTR lengthening (ΔPDUI > 0.1, P-value < 0.05) and 169 genes with 3′UTR shortening (ΔPDUI < − 0.1, P-value < 0.05). The finding that DN glomeruli had significantly more 3′UTR lengthened genes (95%, 2,835/3,004) than shortened ones (5%, 169/3,004) was further confirmed by QAPA analysis, which revealed that 3,340 and 129 genes were lengthened (ΔDPAU > 10, P-value ≤ 0.05) and shortened (ΔDPAU < − 10, P-value ≤ 0.05) in 3′UTR, respectively (Fig. 1c, d, bottom). The DN-associated lengthening events were predominantly within the length of 200–300 bp fragment sequences (Fig. 1e).
Fig. 1
Global lengthening of 3′UTRs in DN identified from RNA-seq data and qRT-PCR verification. a The workflow for the identification of dynamic APA events from glomerular RNA-seq data. b PCA analysis of PDUI (top) and DPAU (bottom) score clearly separated DN patients from control subjects. c Scatter plots of median PDUI (top) or DPAU (bottom) scores between DN and control for each gene. Dashed lines represent ± 0.1 cutoffs for PDUI and ± 10 cutoffs for DPAU, respectively. The different 3′UTR isoforms resulted from the usage of distal PAS (red) or proximal PAS (blue) in DN were colored. d The volcano plots of 3ʹUTR lengthening (red) and shortening (blue) genes. The cut off value was ∆PDUI (DN-C) = ± 0.1 and -log10 (P-value) = 1.301 (1.301 corresponds to P-value = 0.05) for DaPars algorithm (top), ∆DPAU (DN-C) = ± 10 and -log10 (P-value) = 1.301 for QAPA algorithm (bottom). e The histogram showed the number of genes with 3ʹUTR lengthening and shortening due to APA. f Two representative RNA-seq tracks of dynamic APA-regulated genes (CYB5R1, PDLIM1) to highlight the 3′UTR coverage differences between DN and control samples. Purple track represented DN and blue track represented control. The red box indicated the different part of distal 3ʹUTR. g–h, The quantification of PDUI (g) and DPAU (h) changes for CYB5R1 (top) and PDLIM1 (bottom) between DN and control samples. i qRT-PCR analysis to verify the changes of distal and proximal PASs usage of CYB5R1 and PDLIM1 between DN and control. Schematic diagrams of the primer pair design were illustrated in left panel. The increased usage of distal PASs (L/T) in DN compared with control were quantified (middle and right panel). The data represented the mean ± SEM of six independent experiments
The visualization of RNA-seq tracks of the representative dynamic APA-regulated genes, such as CYB5R1, PDLIM1, PDCD6, MYOF and CFH, confirmed the longer 3′UTRs tracks mainly in DN glomeruli but not in control. Compared with DN samples, the distal 3′UTR tracks in control renal samples almost disappeared, but the proximal 3′UTR tracks remained unchanged (Fig. 1f; Additional file 2: Fig. S2b). These differential 3′UTRs tracks between DN and control were consistent with the ΔPDUI and ΔDPAU scores calculated by DaPars and QAPA algorithms (Fig. 1g–h; Additional file 2: Fig. S2c, d), respectively. To verify the alteration of 3′UTR length underlying DN, qRT-PCR analysis was conducted using CYB5R1 and PDLIM1 genes, with the primers for the two isoforms being designed based on the 3’UTR sequence differences (Fig. 1i, left). The short primer was common to both APA isoforms (shorter and longer, S + L) and the long primer was unique to the longer isoform (L). The qRT-PCR results clearly showed a greater usage percentage of the distal PAS for CYB5R1 and PDLIM1 genes in DN glomeruli compared to that of the control ones (Fig. 1i, middle and right). Collectively, these results suggested that DN glomeruli exhibited a genome-wide APA and nearly 95% of APA-regulated genes used distal poly(A) sites in 3′UTRs. Further GO enrichment results demonstrated that those genes with 3′UTRs lengthening were mainly enriched in inflammation-related biological processes such as endoplasmic reticulum stress pathways, NF-κB signaling, autophagy, cell-cell adhesion and vesicle-mediated transport, etc. The representative 3′UTR lengthened genes enriched in these biological processes were listed in Table 1.
Table 1 The significant biological processes enriched by 3′UTR lengthened genes in DN The existence of post-transcriptional regulatory mechanisms in DN glomeruliGiven that APA is a critical part of post-transcriptional regulation, we next determined the post-transcriptional regulation in DN. First, we obtained unbiased glomerular proteomics profiles in parallel with RNA-seq-based transcriptomics to compare the landscape differences between proteomics and transcriptomics. A total of 15,593 mRNAs and 3192 proteins with high confidence were identified, respectively (Additional file 5: Table S4.1 and S4.2). Both t-SNE (t-distributed Stochastic Neighbor Embedding) and unsupervised HC (Hierarchical Clustering) analysis demonstrated a clear separation between DN patients and controls both in transcriptomics (Fig. 2a, top; Additional file 2: Fig. S3a) and proteomics (Fig. 2a, bottom; Additional file 2: Fig. S3c). Further differential expression analysis identified 2,052 mRNAs (FDR ≤ 0.01, fold change > 2), among which 1,547 mRNAs were upregulated and 505 ones were downregulated in DN (Fig. 2b, top; Additional file 2: Fig. S3b; Additional file 5: Table S4.3). Meanwhile, a total of 1047 significantly differentially expressed proteins were identified, with 931 upregulated and 116 downregulated in DN (Fig. 2b, bottom; Additional file 2: Fig. S3d; Additional file 5: Table S4.4). Many well-known DN-associated genes, such as TFGB1, HSPG2, FN1, COL14A1, COL6A2, C3, SYNPO, NPHS1 and NPHS2 [4, 5] displayed significant differential expression between DN and control glomeruli in both mRNA and protein levels, indicating the reliability of the datasets. (Additional file 2: Fig. S3e and S3f). Further GO enrichment analysis demonstrated that both the upregulated mRNAs and proteins were significantly enriched in complement activation, immune response, inflammation, NF-κB signaling, collagen catabolic process, cell adhesion, extracellular matrix organization, translation, mRNA catabolic process, and leukocyte migration etc. The downregulated mRNAs and proteins were clustered in various metabolic processes involving glucose, fatty acids and amino acids, as well as in some oxidation-reduction processes (Fig. 2c).
Fig. 2
Higher protein translation in DN glomeruli compared to controls. a The t-SNE analyses of transcriptomic (top) and proteomic (bottom). Red nodes represented DN patients and blue nodes represented controls. b The volcano plot of transcriptomic (top) and proteomic (bottom). The differentially expressed genes (2-fold, FDR ≤ 0.01) were highlighted with red (upregulated) and blue (downregulated). c Two-dimensional annotation of biological process (BP) enrichment analysis of differential proteins and mRNAs. The signed -log10 (P-value) values of the BP enrichment at the protein and mRNA level are indicated in the x and y axes, respectively. Red nodes represented the BPs enriched by upregulated mRNAs and proteins, whereas blue nodes represented the BPs enriched by downregulated mRNAs and proteins. d Venn diagram showed the number of matched mRNA-protein pairs (top) as well as the overlap of significantly differentially expressed (DE) mRNA and protein (bottom). e Protein-per-mRNA FC ratio analysis illustrated the fold-change discordance between mRNA and the corresponding protein. The value of FC ratio was log10 transformed. The FC ratio value for 2961 matched genes (left) and 1809 unchanged genes (middle) were log-normally distributed; The FC ratio analysis for 1152 differentially expressed genes identified two populations of extreme values (right). f The violin plot showed the difference in protein-per-mRNA FC ratio between the differentially expressed genes and the non-differentially expressed genes. P-value was calculated by Mann-Whitney U test. g–h The mRNA (left) and protein (right) scatterplots for CYBR1 (g) and PDLIM1 (h) between DN and control samples-h The mRNA (left) and protein (right) scatterplots for CYBR1 (g) and PDLIM1 (h) between DN and control samples
Subsequently, we integrated the transcriptomics and proteomics profiles to systematically explore expression differences between proteins and their corresponding mRNAs. A total of 2,961 matched mRNA-protein pairs were selected in our cohort (Fig. 2d, top). The Spearman’s correlation coefficients of expression levels and expression changes (DN/control) for those mRNA-protein pairs were both relatively weak (Additional file 2: Fig. S3g and Fig. S3h). Among the 2961 matched mRNA-protein pairs, 177 genes were differentially expressed at mRNA and protein levels, while 196 mRNAs and 779 proteins were differentially expressed at mRNA or protein levels (Fig. 2d, bottom; Additional file 2: Fig. S3i). To compare the FC (fold change, DN/C) discordance between mRNA and their corresponding protein, the protein-per-mRNA FC ratio for each gene was calculated. The analysis results demonstrated that, for 2961 mRNA-protein matched genes, the median value of FC ratio was 0.15 (Fig. 2e, left; Additional file 6: Table S5.1). Among them, the median FC ratio of 1809 unchanged genes was 0.10 (Fig. 2e, middle; Additional file 6: Table S5.2). However, for 1152 differentially expressed genes, their FC ratio showed a bimodal pattern, with median values of 0.45 and − 0.29, respectively (Fig. 2e, right; Additional file 6: Table S5.3), and the proportion of genes with positive FC ratio was significantly greater than that with negative FC ratio. In addition, there was also a significant FC ratio discrepancy (P-value = 1.80 × 10− 112, Mann-Whitney U-test) between the differentially expressed genes and the unchanged ones (Fig. 2f). Interestingly, the DN-associated genes CYB5R1, PLIM1, PDCD6, MYOF and CFH, all possessing a lengthened 3′UTR in DN glomeruli, exhibited significantly increased protein levels but unchanged mRNA levels compared to that in control samples (Fig. 2g–h; Additional file 2: Fig. S4).
Contribution of APA-induced 3′UTR lengthening to the increase of protein translation of DN-associated genesGiven that the cis-acting elements in 3′UTRs sequence control crucial post-transcriptional regulation processes [26, 38, 39], we then investigated whether the APA-induced global lengthening of 3′UTRs in DN glomeruli would affect protein translation. The combination of APA results and omics data indicated that a higher number of 3ʹUTR lengthened genes were upregulated at proteins levels compared with mRNA levels (Fig. 3). DaPars revealed that 31.4% (297/947) of the 3ʹUTR lengthened genes increased at protein levels, but just 5.9% (56/947) of them increased at mRNA levels (Fig. 3a, Additional file 2: Fig. S5a). Similarly, the QAPA algorithm demonstrated that 30.8% (306/993) of the 3ʹUTR lengthened genes were upregulated at protein levels, whereas 4.6% (46/993) of them were upregulated at mRNA levels (Fig. 3b; Additional file 2: Fig. S5b). It was suggested that APA-mediated 3ʹUTR lengthening was associated with the increase of protein translation in DN. Furthermore, more genes showed a significant positive correlation between the proportion of distal PAS usage and their corresponding protein abundance, which may be attributed to the fact that the longer 3ʹUTR contains more additional regulatory elements that can enhance protein translation (Fig. 3c).
Fig. 3
The APA-induced 3′UTR lengthening promoted protein translation. a Scatterplots between ΔPDUI (DN-C) and expression changes in proteins (left) and mRNAs (right) levels for the mRNA-protein matched genes with significantly longer (ΔPDUI > 0.1, P-value ≤ 0.05) and shorter 3′UTRs (ΔPDUI < − 0.1, P-value ≤ 0.05). The genes were significantly upregulated (red) or downregulated (blue) (2-fold) in DN, respectively. b Scatterplots between ΔDPAU (DN-C) and expression changes in proteins (left) and mRNAs (right) levels for the mRNA-protein matched genes with significantly longer (ΔDPAU > 10, P-value ≤ 0.05) and shorter 3′UTRs (ΔDPAU < − 10, P-value ≤ 0.05). c The density plots of the statistically significant spearman correlation coefficient (P-value < 0.05) between PDUI (left) or DPAU (right) score and protein abundance for the mRNA-protein matched genes. The dashed lines represented the median value of positive (red) and negative (blue) correlation coefficient. d, e The experimental validation of the APA-mediated 3ʹUTR lengthening in DN increasing protein translation. Schematic diagrams of the pcDNA3.1(+)-CYB5R1-LUTR and pcDNA3.1(+)-CYB5R1-SUTR plasmid constructs were illustrated in d (left panel). The qRT-PCR (d, right panel) and western blot (e) of CYB5R1 in human podocytes under hyperglycemia were performed after transfecting cells with a plasmid expressing long or short 3′UTR isoform. The data represented the mean ± SEM of three independent experiments.
Next, to experimentally verify the influence of APA events on protein translation, we overexpressed different isoforms of CYB5R1 mRNA with short or long 3ʹUTRs in human podocytes stimulated with high concentration of glucose (HG), and then detected the differences in mRNA and protein expression. As depicted in Fig. 3d (left), we cloned the CYB5R1 coding DNA sequence (CDS) into pcDNA3.1(+) plasmid and fused it with either long or short 3′UTR. Podocytes transfected with various CYB5R1-expressing plasmids were then exposed to high glucose. The qRT-PCR analysis of CYB5R1 after plasmid transfection indicated that the mRNA levels of CYB5R1 isoforms with short or long 3ʹUTRs were similar (Fig. 3d, right). However, western blot analysis showed that the long 3ʹUTR isoform expressed significantly more CYB5R1 protein than the short isoform and control pcDNA3.1(+) plasmid under hyperglycemic conditions (Fig. 3e). Briefly, these results corroborated the conclusion that APA-mediated 3ʹUTR lengthening can contribute to enhancing protein translation in DN.
Potential regulatory mechanisms of poly(A) site selection and translation enhancement in DNA growing number of core polyadenylation factors have recently been identified as regulators in PAS selection [13, 33, 40]. To determine the APA regulators behind DN, we observed the protein expression changes of 22 important APA regulators according to our proteomic data (Additional file 2: Fig. S5c). Among these APA-regulatory factors, several factors promoting the selection of distal PAS were expressed at higher levels in DN compared to control subjects (Additional file 2: Fig. S5c). For example, CFIm complex (Cleavage Factor Im complex), a heterodimer consisting of CFI25/CPSF5/NUDT21 and CFI68/CPSF6 or CFI59/CPSF7, were reported to preferentially bind to distal PAS, and the upregulation of CFIm subunits promoted distal PAS usage [41, 42]. Other factors, such as SNRNP70 (as a component of the multi-subunit RNP U1 snRNP) and polyadenylate-binding protein 1 (PABPC1), exhibited similar effects on increasing distal PAS site usage, and upregulation of these factors led to polyadenylation at distal sites [43,44,45]. In contrast, CstF64/CSTF2 has an opposite function, with CstF64 reduction enhancing the usage of distal PAS sites [16, 33]. As shown in Fig. 4a, we found that the abundance of CFIm factors, SNRNP70 and PABPC1 at the protein level was strongly enhanced in DN patients, whereas these proteins were nearly absent in control subjects. However, the protein level of CstF64 was lower in DN, although the difference was not significant between DN patients and controls (Fig. 4a). These results suggested that CFIm factors, SNRNP70 and PABPC1, but not CstF64, served as potential master regulators of distal PAS usage in DN. These APA regulators may be the potential therapeutic targets for DN patients.
Fig. 4
The potential regulation mechanisms of PAS selection and translation enhancement in DN. a The protein scatterplots of the representative polyadenylation factors (NUDT21, CPSF6, SNRNP70, PABPC1 and CstF64) between DN patients and control subjects. b The number of genes gaining RBP-binding sites due to the lengthening of 3′UTR. 77% 3′UTR lengthened genes have gained at least one predicted RBP binding site. c The schematic of 3′UTR lengthening increasing gene translation in DN. Genes such as CYB5R1 and PDLIM1 preferred the use of proximal PAS under normal conditions. In DN, upregulated polyadenylation factors (NUDT21, CPSF6, SNRNP70, and PABPC1) resulted in higher usage of distal PASs and thus increased the abundance of the isoform with longer 3′UTR, which produced more protein through gaining more RBP binding sites
By choosing distal PASs in DN, APA-mediated 3′UTR lengthening could provide more binding sites for miRNA or RBPs. Due to the binding of miRNA exerted translation repressive effect, we speculated that RBPs, but not miRNAs, were the master regulators in DN. Meanwhile, previous studies have demonstrated that protein translation could be regulated by RBPs [46, 47]. Therefore, to explore whether the increased protein translation of APA-induced 3′UTR lengthening genes in DN was due to the enhanced interactions with RBPs, we first calculated the median difference of FC ratio (log2△FC ratio) between RBPs-bound and RBPs-unbound genes. The RBP binding information was extracted from the POSTAR2 database, which is the largest post-transcriptional regulation database including RBP-binding sites derived from various CLIP-seq datasets [48]. The results revealed that among 169 RBPs, 26 RBPs were identified to significantly improve protein translation in DN (log2∆FC ratio > 0, P-value < 0.05), including NOP56, FUBP3, FBL, EWSR1, FXR1, TAF15 and HNRNPA1 (Additional file 7: Table S6.1-6.2). According to our proteomic data, 15 RBPs were detected, 14 of which were upregulated or unchanged at the protein level in DN (Additional file 7: Table S6.3). Many of the those RBPs that promote translation in DN are well-known translational regulatory genes, such as NOP56, FBL, and FUBP3, which have been reported to improve protein translation in various biological processes [24, 49, 50]. These results supported the view that RBPs can regulate the protein translation of DN-associated genes by interacting with cis-acting elements in 3′UTR sequence.
Next, we evaluated whether the obtaining of more RBP binding sites from 3′UTR lengthening could enhance protein translation. To determine the global patterns of APA-mediated increases in RBP binding sites, we searched for the gained RBP binding sites for all 3′UTR lengthened genes. The results demonstrated that ~ 80% (726/943) genes with a lengthened 3′UTR in DN gained at least one predicted RBP binding site compared to control 3′UTR, implying that RBPs may play a critical role in increasing the protein translation of DN-associated genes with a lengthened 3′UTR in DN (Fig. 4b). The representative DN-associated genes with improved protein translation, which bound to the selected translational enhancer RBPs through an extended 3′UTR sequence, were listed in Additional file 7: Table S6.3. Consequently, our results suggested that the DN-associated genes with APA-regulated 3′UTR lengthening had higher protein translation, because they possessed more RBP-binding sites and were more likely to be regulated by certain translational enhancer RBPs (Fig. 4c).
留言 (0)