
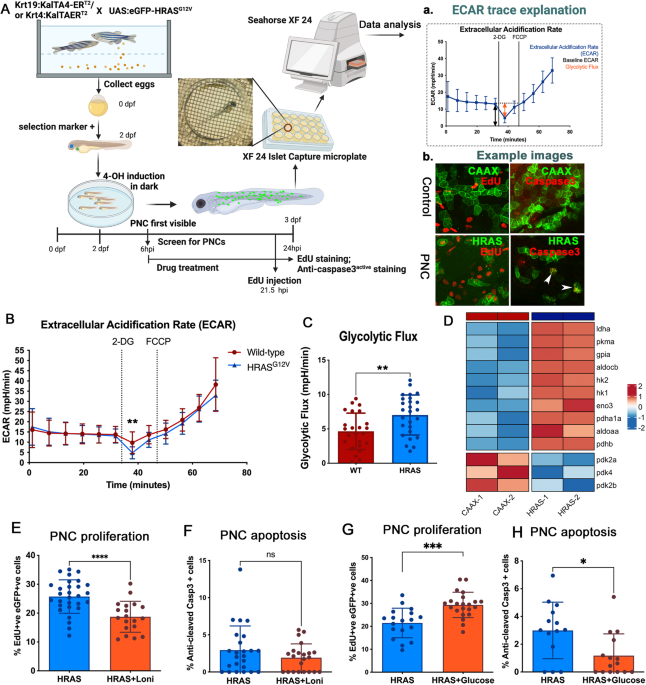
記住我


Since the c-MYC pathway has been demonstrated to significantly contribute to HCC oncogenesis, we evaluated the effects of AR-V7 expression on the c-MYC-driven hepatocarcinogenesis in vivo. To generate the mouse model, we stably transfected the c-MYC expression vector pT3-c-MYC with the AR-V7 expression vector pT3-AR-V7 (hereby designated as hep-c-MYC/AR-V7) or without pT3-AR-V7 (designated as hep-c-MYC) into mouse hepatocytes using the hydrodynamic tail-vein injection technique (Fig. 1B). We also generated the hep-c-MYC/AR-FL mice and control mice similarly by hydrodynamic injection of the pT3-c-MYC and pT3-AR-FL or pT3-EF1α empty vector, respectively. The expressions of c-MYC, AR-V7, and AR-FL in the developed tumor foci were confirmed by immunohistochemistry at 20 days post-injection (dpi) (Fig. 1C). When AR-V7 alone was injected in the adult mice, no tumor was observed in their livers up to 20 dpi. However, AR-V7 expression was observed in selected hepatocytes at 3 dpi but was barely detectable at 20 dpi (Supplementary Fig. 4).
At 20 dpi, the overall tumor volume in the hep-c-MYC/AR-V7 mice was larger than hep-c-MYC mice regardless of the sexes as indicated by increased liver-to-body weight ratio (Fig. 1D, E; Supplementary Fig. 3). Similarly, the relative tumor area was larger in hep-c-MYC/AR-V7 mice than hep-c-MYC mice in both sexes (Fig. 1F and Supplementary Fig. 3). These results suggest that AR-V7 potentiated the c-MYC-driven hepatocarcinogenesis in a ligand-independent manner.
Histopathologic analyses of hematoxylin-eosin (H&E) stained sections revealed that tumors developed in hep-c-MYC mice were moderately steatohepatitic, contained small droplets of fat in cancer cells (Fig. 1G, c-MYC). On the other hand, tumors developed in hep-c-MYC/AR-V7 mice were morphologically heterogenous, with steatohepatitic and non-steatohepatitic foci regardless of the sexes (Fig. 1G, c-MYC/AR-V7). Cells in non-steatohepatitic tumor foci had scant cytoplasm without droplets of fat (Fig. 1G, c-MYC/AR-V7).
On the other hand, the relative tumor area in hep-c-MYC/AR-FL male mice were morphologically larger than that in hep-c-MYC male mice (Fig. 1F and Supplementary Fig. 3). In addition, histopathologic analyses showed that non-steatohepatitic tumor foci were developed in male c-MYC/AR-FL mice similar to hep-c-MYC/AR-V7 mice (Fig. 1G, c-MYC/AR-FL). In contrast, non-steatohepatitic tumor foci were not developed in female c-MYC/AR-FL mice and relative surface tumor area was not obviously different from that of female hep-c-MYC mice (Fig. 1F and Supplementary Fig. 3). These results indicate that AR-FL could also exert mild effects, albeit at much lower levels than those for AR-V7, on c-MYC-driven hepatocarcinogenesis in males but not females, suggesting a male-biased exacerbation of the resulting HCC between the sexes.
Transcriptome analyses revealed the biological processes that were synergistically altered by c-MYC and AR-V7The c-MYC plays significant roles in many biological processes, including proliferation, protein and ribosomal biosynthesis, metabolism, immune surveillance, cell differentiation, cell adhesion, and senescence. When it is aberrantly overexpressed/activated, it amplifies the biological processes toward oncogenesis, such as gene instability, accelerated cell proliferation, metabolism, and angiogenesis, resulting in cancer development [4]. To explore the likely mechanisms by which AR-V7 potentiates the c-MYC-driven hepatocarcinogenesis, we performed a gene expression profiling of liver cancer developed in hep-c-MYC and hep-c-MYC/AR-V7 mice at 20 dpi by RNA-seq transcriptome analyses. Liver samples from mice at 20 dpi injected with an empty vector alone were used as controls. Since AR-V7 and c-MYC cooperatively promoted liver cancer regardless of the sexes, we identified the genes that were commonly altered in male and female mice. The results showed that the expression patterns of 2930 genes were differentially altered by c-MYC overexpression (Supplementary Table 4). Out of these, 2691 genes were not further affected by AR-V7 co-expression (class-C, yellow in Fig. 2A). AR-V7 co-expression potentiated the c-MYC-mediated changes of 17 genes (class-D, red in Fig. 2A), completely (class A) and partially (class-B) repressed 181 and 41 c-MYC-regulated genes respectively (green and blue in Fig. 2A). In addition, expression patterns of 243 genes were affected by AR-V7 co-expression but not by c-MYC alone (class-E, light red in Fig. 2A), and hence could be attributed to AR-V7 effects under such conditions. The functional annotation enrichment analysis using the DAVID bioinformatics resources [31] revealed that various c-MYC-regulated biological processes, including proliferation, protein and ribosomal biosynthesis, and gene instability were significantly affected, while metabolism and other pathways were further altered by AR-V7 co-expression at various levels (Fig. 2B). Processes in metabolism were mostly affected by the AR-V7 co-expression (Fig. 2B, right panel). An enrichment analysis that focused on the genes potentiated or promoted by AR-V7 co-expression (dark-red and light-red areas in Fig. 2A) suggested that major portions of the genes involved in lipid metabolism and steroid/sterol metabolism were significantly affected by AR-V7 (Fig. 2C).
Fig. 2: Effect of the co-expression of AR-V7/AR-FL on the gene expression profiles of the c-MYC-driven liver cancer.
A Number and classification of the differentially expressed genes (DEGs) in mouse HCC promoted by c-MYC alone (c-MYC) or c-MYC and AR-V7 co-expression (c-MYC/AR-V7) at 20 dpi (class A-E). AR-V7 affected only subsets of c-MYC-mediated DEGs. B Results of DAVID functional enrichment analyses for the DEGs in the mouse HCC samples of hep-c-MYC (common between male and female), hep-c-MYC/AR-FL (male), or hep-c-MYC/AR-V7 (common between male and female). Selected categories of the c-MYC-regulated processes are indicated on left. Right panel indicates the portions of genes in each biological process affected by AR-V7, with the most effects being in those in metabolism. C Metabolic processes promoted by AR-V7 (dark blue) and shared with AF-FL (light blue) co-expression with c-MYC, respectively.
The macroscopic analyses showed that AR-FL co-expression moderately exacerbated the c-MYC-driven liver cancer in male mice, but not in female mice (Fig. 1F). Hence, we further investigated the gene expression profiles and enriched biological processes in the male hep-c-MYC/AR-FL liver cancer. The results showed that the AR-FL could share portions of the gene sets in metabolism processes affected by the AR-V7 co-expression (Fig. 2C, right panel), suggesting that AR-FL exacerbates, at a reduced level, certain c-MYC-driven hepatocarcinogenic processes in male mice similar to those of AR-V7.
To evaluate the effects of AR-V7 on c-MYC-driven hepatic oncogenesis, we examined specific genes, i.e., B4galnt1/G4GALNT1, Ffar4/FFAR4, and Il1rl2/IL1RL2, that was further upregulated by AR-V7 co-expression, as indicated in class-D genes (Fig. 2A, red). The corresponding survival patterns in HCC patients with high and low expression of the respective class-D genes in the TCGA database were analyzed. These genes were reported in the literature to be associated with oncogenesis of various human cancers. The B4galnt1 gene encodes the enzyme beta-1,4-N-acetylgalactosaminyl transferase, involved in the biosynthesis of complex gangliosides. Various studies demonstrated that B4GALNT1 was involved in the progression of different cancer types including oral squamous cell carcinoma and lung adenocarcinoma [32, 33]. Quantitative RT-PCR analysis showed that it was upregulated by c-MYC but was further elevated by AR-V7 co-expression in both male and female HCC samples (Fig. 3A, far left panel). Its expression level was negatively correlated with the survival ratio in HCC patients (Fig. 3B, far left panel). Several other genes, including the free fatty acid receptor 4 (Ffar4), and interleukin 1 receptor like 2 (Il1rl2), also showed similar expression patterns in our mouse model and their high levels of expression correlated with poor clinical outcomes in HCC patients (Fig. 3A, B). FFAR4 has been demonstrated to promote epithelial-mesenchymal transition, cell proliferation/migration, and drug resistance in various cancer types [34]. IL1RL2 (also known as Interleukin 36 receptor, IL36R) is involved in tissue fibrosis and metastatic potential in breast and colon cancers [35, 36]. Our results showed that AR-V7 could upregulate the expression of these pro-oncogenic genes, and directly or indirectly potentiate the c-MYC-related oncogenic processes, resulting in exacerbation of the c-MYC-driven HCC. AR-FL co-expression showed moderate increases in the expression of these pro-oncogenic genes (Fig. 3A, B) and could contribute similarly to the exacerbation of the c-MYC-driven liver cancer development in males, but at a much-reduced level. We surmise that such similarities could contribute to the relatively milder exacerbation of c-MYC-driven hepatocarcinogenic processes in hep-c-MYC/AR-FL male mice than those of hep-c-MYC/AR-V7 mice. On the other hand, genes affected at higher levels and/or unique to AR-V7 actions could be responsible for the high exacerbation effects in c-MYC-driven hepatic oncogenesis in the hep-c-MYC/AR-V7 mice.
Fig. 3: Examples of three genes upregulated by AR-V7 co-expression in the c-MYC-driven mouse HCC models and the corresponding survival patterns in HCC patients.
A Quantitative RT-PCR for expressions of B4galnt1, Ffar4, or Il1rl2, in respective sample groups. The Y axis indicates the expression levels relative to Gapdh. Statistical significance by one-way ANOVA with Tukey’s multiple comparisons test; *p < 0.05; **p < 0.001; ***p < 0.0001. Error bars indicate mean ± SEM. B Kaplan–Meier survival plots of HCC patients in TCGA datasets for the indicated genes. Red line indicates high expressor and blue line indicates low expressor, respectively. Log-rank test P value is indicated.
The tumor suppressor Claudin 7 is a potentially key target of AR-V7 in the hep-c-MYC/AR-V7 HCCWhile c-MYC plays pivotal roles in cell proliferation and cancer development, it also increases the sensitivity to apoptosis in non- and pre-malignant cells [37, 38]. Further, hepatocytic c-MYC overexpression promoted liver cancer depending on the developmental context, and adult mice showed a longer latency to develop cancer than newborn mice due to activation of the tumor suppressor p53 [11]. Hence, we speculated that AR-V7 might also reduce the c-MYC-mediated anti-proliferative pathways in the transfected hepatocytes, in addition to potentiating the c-MYC-mediated oncogenic pathways. By exploring the transcriptome data of the 222 c-MYC downstream genes whose expression levels were partly (41 genes) or completely (181 genes) repressed by AR-V7 co-expression (green and blue areas in Fig. 2A and Supplementary Table 4), we found that the tumor suppressor gene Claudin 7 (Cldn7) was upregulated more than 40 folds in hep-c-MYC cancer compared to control liver, whereas such upregulation was reversed in the hep-c-MYC/AR-V7 cancer in both male and female mice (Fig. 4A). Such differential expression was confirmed by quantitative RT-PCR analysis (Fig. 4B). Noteworthy, the c-MYC-induced Cldn7 upregulation was partly repressed by the AR-FL-co-expression in male mice, but not in female mice (Fig. 4B). These results suggest that c-MYC upregulation of the Cldn7 tumor suppressor gene is completely and partially repressed by AR-V7 and AR-FL in a ligand-independent and ligand-dependent manner respectively.
Fig. 4: AR-FL and AR-V7 differently suppressed a tumor suppressor gene Cldn7 that was upregulated by c-MYC overexpression in mouse HCC models.
A RNA-seq transcriptome analysis showed that 150 genes that were upregulated by c-MYC were repressed by AR-FL and/or AR-V7 co-expression; and 31 genes that were downregulated by c-MYC were upregulated by AR-FL and/or AR-V7 co-expression in both male (left) and female (right) mice at 20 dpi respectively. Each blue line represents differential expression levels (fold change in reference to control, Y axis) in the tumors of the respective combination of injected gene(s), X axis. Red lines highlight the differential expression levels of Cldn7 in respective samples. B Quantitative RT-PCR for expressions of Cldn7 in respective groups at 20 dpi. Expression of Cldn7 in male mice of c-MYC/p53KO (hep-c-MYC/p53KO) tumor at 11 dpi (pink bar), indicating that Cldn7 was repressed in p53-deficient conditions and hence was likely regulated by p53 [70–72]. C Expression of p53 in respective mouse groups in male and female, indicating p53 was upregulated by c-MYC but not significantly affected by AR-FL or AR-V7 co-expression. D Immunofluorescent images of c-MYC (red), Cldn7FLAG (green), and DNA (blue) in the liver of hep-c-MYC/p53KO/Cldn7FLAG mouse at 2 dpi, showing co-expression of the respective transgenes in the same cells. Scale bar = 50 µm E, Macroscopic phenotypes of the livers in male mice at 11 dpi injected with the expression vectors of c-MYC (c-MYC) (n = 5), c-MYC under p53-deficient condition (c-MYC/p53KO) (n = 6), or c-MYC and Cldn7FLAG under p53-deficient condition (c-MYC/p53KO/Cldn7FLAG) (n = 5). F Liver to body weight ratio of the samples presented in E, indicating that the exacerbation of c-MYC-driven HCC under p53-deficient conditions was completely reversed by co-expression of Cldn7FLAG. Statistical significance by one-way ANOVA with Tukey’s multiple comparisons test for B, C, and F; *p < 0.05; **p < 0.001; ***p < 0.0001. Error bars indicate mean ± SEM.
Claudin 7 is a member of the claudin family that is involved in wide variety of biological processes including tight junction formation, cell polarity, signal transduction, transcription regulation, and mRNA stability [39, 40]. Recent studies revealed that the human CLDN7 is upregulated by the p53 signaling pathway and suppresses cell proliferation in various cancer types [41, 42]. Hence, we investigated further the expression and function of Cldn7 in the c-MYC-driven HCC mouse model. First, to explore the correlation between p53 and Cldn7 genes in the hep-c-MYC cancer, we analyzed the effect of the p53-deficiency on the Cldn7 expression in the tumors promoted by the c-MYC overexpression. When c-MYC was overexpressed under the p53-deficient condition (hep-c-MYC/p53KO) by co-injection of pX330-p53, the CRISPR base p53 knockout vector [27], cancer developed rapidly, as compared to those promoted by c-MYC alone. The pX330-p53 is a single plasmid vector capable of inactivating/deleting the p53 gene in the cells via the CRISPR targeting mechanisms. The inactivated p53 gene produces non-functional transcripts (confirmed as the dominant transcripts by sequencing, Supplementary Fig. 5), distinguishable from the wild-type transcript. The tumor size reached the end-point criteria at 11 dpi. A quantitative RT-PCR analysis showed that the expression level of Cldn7 in hep-c-MYC/p53KO cancer (n = 4, male mice) at 11 dpi was significantly lower than that of hep-c-MYC cancer at 20 dpi (Fig. 4B, c-MYC/p53KO). These results suggest that the Cldn7 gene is upregulated by c-MYC in a p53-dependent manner, thereby confirming that Cldn7 is a target of p53 regulation [41, 42]. Although p53 was upregulated in the hep-c-MYC liver cancer samples (Fig. 4C), the AR-V7 co-expression did not significantly affect the p53 expression level (Fig. 4C), suggesting that AR-V7 might directly suppress the Cldn7 expression downstream of p53.
Next, we evaluated the impact of Cldn7 expression on the c-MYC-driven HCC development under p53-deficient conditions in vivo using the hydrodynamic tail-vein injection system. An immunofluorescence analysis showed that c-MYC was localized in the nuclei, the FLAG-tagged Cldn7 (Cldn7FLAG) was localized in cytoplasm, and nucleoli in the hydrodynamically transfected hepatocytes at 2 dpi of pT3-c-MYC, pT3-Cldn7FLAG and pX330-p53 (hep-c-MYC/p53KO/Cldn7FLAG) (Fig. 4D), confirming the co-expression of these two transgenes in the same cells. The co-expression of Cldn7FLAG significantly diminished the hep-c-MYC/p53KO cancer development, and the overall tumor volume in the hep-c-MYC/p53KO/Cldn7FLAG mice was equivalent to that of hep-c-MYC mice at 11 dpi (Fig. 4E, F). These results suggest that c-MYC activation of p53 leads to upregulation of Cldn7 and repression of c-MYC-mediated oncogenesis and overexpression of Cldn7 counteracts the accelerated oncogenesis by c-MYC in the absence of p53 (hep-c-MYC/p53KO). Further, our studies imply that AR-V7 and, to a lesser extent AR-FL in males, could bypass the p53 regulation and directly repress the Cldn7 tumor suppressor, thereby exacerbating the c-MYC-driven hepatic oncogenesis.
留言 (0)