
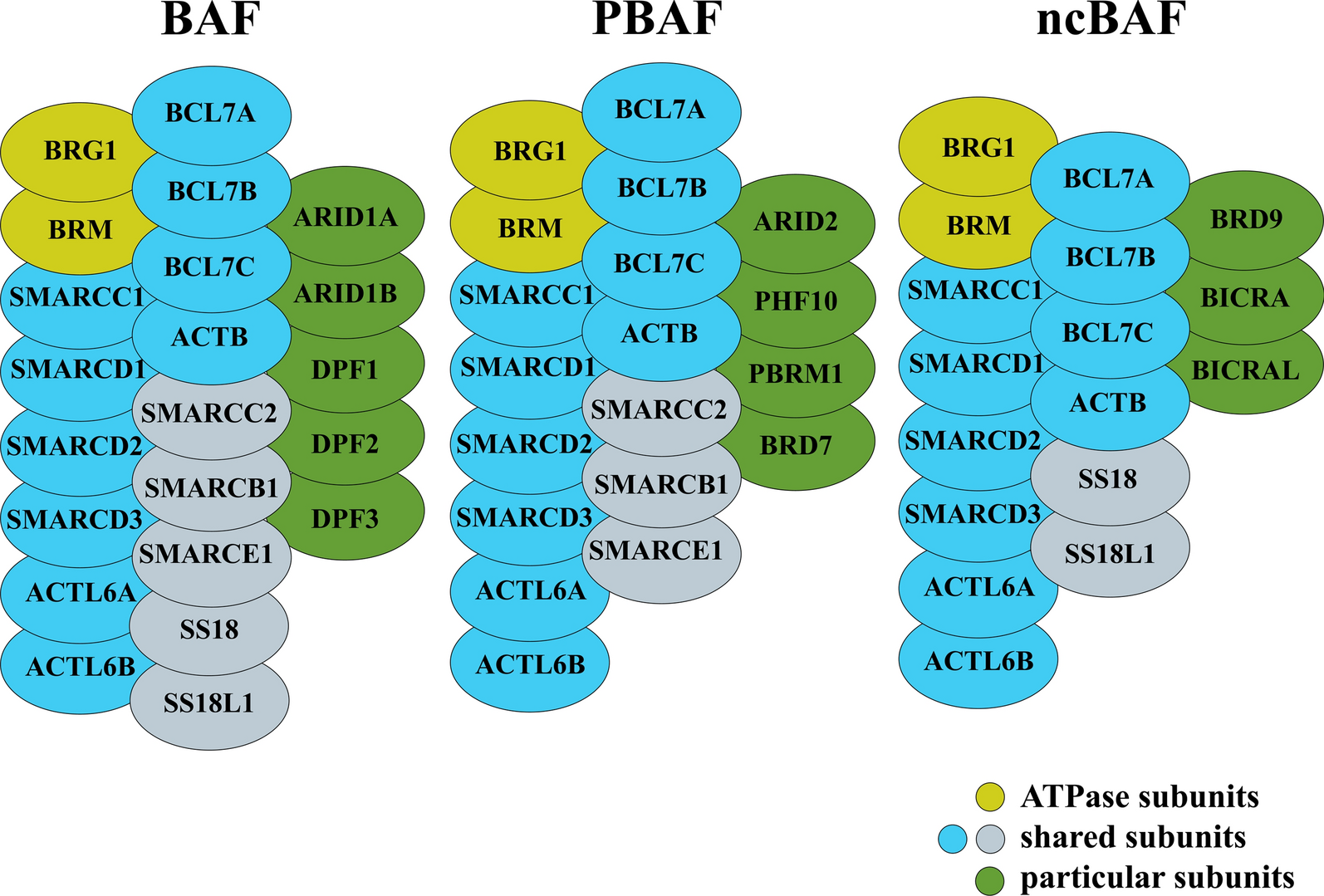
記住我


The key parameter for describing DNA methylation is the methylation probability (MP) of each CpG site, defined as the probability of observing a methylated cytosine on the site. For a CpG site under consideration, its methylation state can be described by a random variable \(X\sim \text (p)\) with outcome “1” representing methylation and “0” otherwise, where \(p\in [0, 1]\) is the MP. Therefore, for a CpG segment involving n sites, the methylation states of the n contiguous sites form a binary random vector \(\varvec=[X_1, X_2, \ldots , X_n]^T\). This random vector represents the “methylation pattern” exhibited on the n-CpG segment. Marginally, \(X_i\sim \text (p_i),\,p_i\in [0, 1],\,1\le i\le n\), but the joint distribution of \(\varvec\) also depends on the correlation among the CpG sites. It has been found that, such a between-site correlation, hereinafter called the spatial correlation, is often nonnegligible [5,6,7] and plays important roles in the analysis of methylation data, such as detecting epigenome-wide association signals [17], discovering regions associated with exposure [18] or with differential/variabe methylation [19], and hunting epigenomic bumps/peaks [20]. We call \(\varvec\) a correlated Bernoulli random vector with parameters \(\varvec\) and \(\varvec\), where \(\varvec=[p_1, p_2, \ldots , p_n]^T\) denotes the mean and
$$\begin \varvec=\left( \begin1&r_&\cdots &r_\\ r_&1&\cdots &r_\\ \vdots &\vdots &\ddots &\vdots \\ r_&r_&\cdots &1\end\right) \end$$
is the correlation matrix, \(r_\in [-1, 1],\,1\le i\ne j\le n\). Note, the range of the correlation matrix is constrained, see details in Prentice [21] and Chaganty and Joe [22]. There are two special methylation patterns that are of particular interest: (1) homogeneous methylation, this refers to the case that \(p_i=p,\,1\le i\le n\), i.e., the n CpG sites share a common MP; and (2) exchangeable (or compound symmetric) correlation, corresponding to \(r_=r\) for \(1\le i\ne j\le n\), i.e., the spatial correlation is fixed, not depending on the distance between the CpG sites.
The MP at single-CpG-site level plays a critical role in differentiating samples (e.g., purified cells), however, for analyzing epigenetic heterogeneity in mixed samples (e.g., bulk tissues), the MP has its limitations, and other characteristics at the read level are required. One of such characteristics is the methylation entropy (ME), defined as a measure of the average level of “information” or “uncertainty” inherent to the methylation pattern exhibited across contiguous CpG sites [11, 12]. Different with the MP which only characterizes the methylation state at each single CpG site, the ME summarizes the joint distribution of the methylation states at multiple sites in a CpG segment. By leveraging read-level stochasticity, the ME can be used to evaluate the variation of the methylation pattern on the CpG segment.
Consider an n-CpG segment fully covered by m sequencing reads. In practice, n is usually chosen to be a small integer, e.g., \(n=4\). This is to guarantee that enough CpG segments can be extracted from real data and the number of sequencing reads in each CpG segment (i.e., m) is sufficiently large. The methylation pattern \(\varvec\) is a binary random vector, taking a total of \(2^n\) possible values. The distribution of \(\varvec\) can be expressed as a function of \(p_i\) and \(r_,\,1\le i\ne j\le n\). Denote the distribution of \(\varvec\) by \(\\}\) such that:
$$\begin q_0= \,& P(\varvec=[0, 0, \cdots , 0, 0, 0]^T), \\ q_1=\, & P(\varvec=[0, 0, \cdots , 0, 0, 1]^T), \\ q_2=\, & P(\varvec=[0, 0, \cdots , 0, 1, 0]^T), \\ q_3=\, & P(\varvec=[0, 0, \cdots , 0, 1, 1]^T), \\&\vdots&\\ q_=\, & P(\varvec=[1, 1, \cdots , 1, 1, 1]^T), \end$$
the ME of the random vector \(\varvec\) is defined straightforwardly from the Shannon entropy [23, 24].
Definition 1The methylation entropy of an n-CpG segment is defined by
$$\begin S=-\sum _^q_i\log (q_i), \end$$
(1)
where \(\\}\) form the probability mass function of the methylation states of the n contiguous CpG sites.
For convenience, base 2 is often chosen for the logarithm so that the ME can be evaluated in the unit of bits. In the absence of spatial correlation, i.e., assuming independence between the CpG sites, an explicit expression of the ME can be derived in terms of the MP.
Corollary 1If the CpG sites on an n-CpG segment are independent, then the methylation entropy
$$\begin S=-\sum _^n\left[ (1-p_i)\log _2(1-p_i)+p_i\log _2(p_i)\right] , \end$$
where \(p_i\) is the methylation probability of the ith CpG site.
ProofDenote the methylation states of the n contiguous CpG sites on the segment by a random vector \(\varvec\). By the assumption of independence between the CpG sites, the distribution of \(\varvec\) can be seen as
$$\begin q_0=\, & P(\varvec=[0, 0, \cdots , 0, 0, 0]^T)=\prod _(1-p_i),\nonumber \\ q_1=\, & P(\varvec=[0, 0, \cdots , 0, 0, 1]^T)=\left[ \prod _(1-p_i)\right] p_n,\nonumber \\ q_2=\,& P(\varvec=[0, 0, \cdots , 0, 1, 0]^T)=\left[ \prod _}(1-p_i)\right] p_, \nonumber \\ q_3=\,& P(\varvec=[0, 0, \cdots , 0, 1, 1]^T)=\left[ \prod _}(1-p_i)\right] p_p_n,\nonumber \\&\vdots&\nonumber \\ q_=\,& P(\varvec=[1, 1, \cdots , 1, 1, 1]^T)=\prod _p_i, \end$$
(2)
where \(N=\\). Plugging in (2) to the ME definition (1), the expression of ME is simplified to \(S=-\sum _^n\left[ (1-p_i)\log _2(1-p_i)+p_i\log _2(p_i)\right]\). That is, under the assumption of independent CpG sites, S is simply the n-bit binary entropy function. □
Given independence between the CpG sites, for the special case of homogeneous methylation, i.e., \(p_i=p\) for all \(1\le i\le n\), Corollary 1 simplifies to
$$\begin S=-n\left[ (1-p)\log _2(1-p)+p\log _2(p)\right] . \end$$
To illustrate the analytical relation in Corollary 1, for a simple example of 2-CpG segment, Fig. 2A shows the ME S as a function of the MPs \(p_1\) and \(p_2\) on the two CpG sites under the independence assumption. In particular, the relation between the ME and the MP for the homogeneous methylation case is shown in Fig. 2B, which is also the curve on the \(p_1=p_2\) plane in Fig. 2A.
Fig. 2
Analytical relation between methylation entropy S and methylation probability p in a 2-CpG segment under independence assumption. A S as a function of \(p_1\) and \(p_2\) on the two CpG sites. B Relation between S and p when \(p_1=p_2=:p\)
Testing for spatial correlationBased on the distribution of the methylation pattern on a CpG segment, we can check the existence of spatial correlation by a Pearson’s \(\chi ^2\) goodness-of-fit test. Under the null hypothesis of no spatial correlation, i.e., \(H_0: r_=0,\,\forall 1\le i\ne j\le n\) or the correlation matrix \(\varvec\) is an identity matrix, the distribution of the methylation pattern is provided in (2). Suppose that a total of m sequencing reads are collected in bisulfite sequencing to cover this segment, and denote the number of reads showing different methylation patterns corresponding to \(\\}\) by \(O_0, O_1, \ldots , O_\). These observed counts \(O_i\) together with their expected values \(E_i,\,0\le i\le 2^n-1\) under \(H_0\) form the following contingency table:
Expected \(E_i\)
\(E_0=mq_0\)
\(E_1=mq_1\)
\(\cdots\)
\(E_=mq_\)
Observed \(O_i\)
\(O_0\)
\(O_1\)
\(\cdots\)
\(O_\)
The chi-square test can be conducted for goodness-of-fit based on a test statistic
$$\begin \chi ^2=\sum _^ \frac, \end$$
with a degree of freedom \(2^n-1-n\) (when \(0<p_i<1,\,\forall 1\le i\le n\), a total of n MP’s need to be estimated, resulting in a loss of n degrees of freedom).
We performed a simulation study (Simulation 1) to evaluate this chi-square test of spatial correlation in terms of empirical type-I error and power. Details of the simulation procedure, evaluation results, and interpretations are provided in the "Results" section.
Relation between methylation entropy and methylation probability with spatial correlation taken into accountIn the presence of spatial correlation, the ME is a function of both the MP and the spatial correlation, and intuitively, one would expect that for fixed MP, the ME behaves as a decreasing function of the spatial correlation because larger correlation reduces the stochasticity inherent to the methylation pattern. Such a conjecture can be confirmed by calculating the distribution of the methylation pattern numerically, given the parameters \(\varvec\) and \(\varvec\) of the correlated Bernoulli distribution. For the convenience of demonstration, here we show the relation of the ME with \(\varvec\) and \(\varvec\) for the following two cases of homogeneous methylation:
(1)ME vs. MP in a 2-CpG segment. Denote the methylation states of the two CpG sites by random variables \(X_1\) and \(X_2\), respectively. Assuming homogeneous methylation, both \(X_1\) and \(X_2\) follow the same distribution of \(\text (p)\). Let \(cor(X_1, X_2)=r\), the joint distribution of \((X_1, X_2)\) can be obtained explicitly as:
$$\begin q_0=\,& P(X_1=0, X_2=0)=rp(1-p)+(1-p)^2, \\ q_1=\,& P(X_1=0, X_2=1)=p(1-p)(1-r), \\ q_2=\,& P(X_1=1, X_2=0)=p(1-p)(1-r), \\ q_3=\,& P(X_1=1, X_2=1)=rp(1-p)+p^2, \end$$
and consequently, by Definition (1) the ME S can be written as a function of p and r. Figure 3A shows this analytical relation between S and p for a 2-CpG segment.
(2)ME vs. MP in an n-CpG segment, \(n>2\). When \(n>2\), the distribution of the methylation pattern cannot be directly derived from the pair-wise correlations, yet the thresholding method in Emrich and Piedmonte [25] can be adopted to bypass this difficulty. The basic idea is to relate the correlated Bernoulli random vector to a latent multivariate normal (MVN) random vector by dichotomization in each dimension, determine the truncation parameters and covariance matrix of the MVN distribution by using the given parameters of the correlated Bernoulli, and calculate the distribution of the methylation pattern from the joint cumulative distribution function (CDF) of the MVN. For the convenience of demonstration, Fig. 3B shows for a 4-CpG segment the analytical relation among ME, MP, and spatial correlation, under exchangeable correlation and homogeneous methylation assumptions.
Fig. 3
Analytical relation between methylation entropy S and methylation probability p in an n-CpG segment, under different settings of spatial correlation parameter r. The 21 curves from top to bottom correspond to \(r=0,\,0.05,\,\ldots ,\,0.95,\,1\). For each r setting, S is shown as a function of p. A: \(n=2\), under homogeneous methylation (i.e., \(p_1=p_2=:p\)). B: \(n=4\) under exchangeable correlation (i.e., \(r_=r,1\le i\ne j\le 4\)) and homogeneous methylation (i.e., \(p_i=p,1\le i\le 4\))
The analytical relation between the parameters can be further validated by its analogous counterpart in terms of statistics. For this purpose, we performed a simulation study (Simulation 2) to illustrate the empirical relation between two statistics, the observed ME (OME) and the mean methylation level (MML), in the simulated data. Denote the simulated methylation data at each CpG segment by an \(m\times n\) binary matrix \(\varvec\), that is, assuming a total of m sequencing reads are collected to fully cover the n-CpG segment. The OME statistic \(\hat\) is defined as:
$$\begin \hat=-\sum _^\frac\log \left( \frac\right) . \end$$
The CpG-site-based ML (i.e., the fraction of methylated cytosines on each CpG site) is the sample mean statistic
$$\begin \bar_j=\frac\sum _^m x_, \quad 1\le j\le n, \end$$
and the MML statistic is simply the overall sample mean across the n-CpG sites. From elementary statistics, the OME and ML are the unbiased estimators of the parameters ME and MP, respectively (MML is also the unbiased estimator of the homogeneous MP). Details of the simulation procedure, results, and interpretations are provided in the "Results" section.
Real data applicationGuided by the theoretical results presented in previous sections as well as the two simulation studies, we applied the proposed method to analyze a published dataset of mouse brain methylome [26]. This dataset includes MethylC-Seq data on mouse frontal cortex during early postnatal, juvenile, adolescent, and adult stages. Our analysis contains three steps: (1) for a given sample, extract all 4-CpG segments under a prespecified depth, (2) calculate the OME and MML statistics for each segment, and draw a scatter plot to show the relation of OME vs. MML, and (3) mark CpG segments which exhibit low OME, non-extreme MML, and strong spatial correlation. As elucidated, the low OME and strong spatial correlation in these segments suggest “bi-modality” in the distribution of methylation pattern, which indicates strong epigenetic control mechanism and can be a potential signal of CSM arising from a mixture of two cell types [16]. We will call such bi-modal distributed CpG segments the bipolar methylated loci.
There are two specific aims, association validation and dynamics demonstration, for this real data application. The first one is to check whether the identified bipolar methylated loci are associated with DMRs. For this purpose, we choose two samples that contain bisulfite sequencing data enriched with neuron and glia cells, respectively. The sequencing reads from these two samples are pooled together to form data of a mixture of two cell types, and bipolar methylated loci are then identified from this mixed cell population. Meanwhile, by comparing the mean methylation levels of the CpG segments obtained from the two samples separately, DMRs are detected between neuron and glia cells. We then check the association (or overlap) between the identified bipolar methylated loci and DMRs to validate whether the proposed method helps discover CSM. Second, for dynamics demonstration, we would like to see how the bipolar methylated loci change over time. This is done by choosing two samples from different stages (e.g., 2-week and 4-week) of the mouse frontal cortex data, and comparing the corresponding bipolar methylated loci identified by Steps (1)–(4) above. This comparison sheds light on the dynamics of bipolar methylated loci during mouse brain development. Details of the real data analysis procedure, results, and discussions are provided in the "Results" section.
留言 (0)