Fish maintenance
All diploid loach used in this study were cultured in the recirculating water system with the temperature of 24–26 °C, pH of 7.0–7.5, and dissolved oxygen (DO) of 6.0–6.5 mg/L. Artemia and tubificidae were used to feed loach larvae and loach fingerlings/adults, respectively. The loach was fed three times a day (8:00, 14:00, and 20:00).
Genome sequencing
The muscle tissues from one diploid male WT loach M. anguillicaudatus were used to extract genomic DNA by using the classic phenol-chloroform method. Quality and quantity of the extracted DNA were assessed using the Nanodrop and Qubit, respectively. And the integrity of the extracted DNA was further evaluated on an agarose gel stained with ethidium bromide. The qualified genomic DNA was then sequenced on the platform of PacBio Sequel sequencing (Pacific Biosciences). Genomic DNA of the loach was sheared to an average size using a g-TUBE device (Covaris, Woburn, MA, USA). The sheared DNA was purified and end-repaired using polishing enzymes and then a blunt end ligation reaction followed by exonuclease treatment was applied to create a SMRTbell template according to the PacBio template preparation protocol. After the construction of the library, Qubit 3.0 and Agilent 2100 were used for quantification and detecting the library size, respectively. The library single-molecule sequencing was then conducted on the PacBio Sequel platform to generate long-read data. Moreover, the genome of loach was further sequenced on the Hi-C platform to obtain chromosome-level genome assembly. For Hi-C library construction, DNA extracted from the loach was fragmented and purified using magnetic beads. A Hi-C library (300–350 bp) was constructed and sequenced on the Illumina HiSeq 4000 platform with 150-bp paired-end reads. For RNA sequencing, the complementary DNA libraries were constructed from various tissues according to the manufacturer’s instructions (NEBNext Ultra RNA Library Prep Kit from Illumina, catalog no. E7530S) and sequenced on the Illumina HiSeq 4000 platform (Additional file 1: Table S6).
Quality control of sequencing data
The different reads including Illumina reads, PacBio reads, and Hi-C reads were quality filtered using different strategies. For Illumina sequencing reads and Hi-C reads, all low-quality reads, duplicated reads, and adapter sequences were removed. For the PacBio reads, the adapter sequences were removed first. Then, any of the reads, which had the content of N exceeding 10% or the number of low-quality base (≤ 5) exceeding 50% of the length of this read, the paired reads were filtered in the single-read sequencing. For Hi-C sequencing data, the low-quality Hi-C reads were filtered by HiC-Pro software [63].
Genome size estimation
The genome size of the loach was estimated using the K-mer method. The 17-mer was chosen for K-mer analysis in this study, and the genome size (G) was estimated based on the following formula: G = k-mer number/k-mer depth, where k-mer number and k-mer depth represented the total number and the peak of depth of the 17-mer, respectively.
Genome assembly and chromosome construction
PacBio reads were corrected, trimmed, and assembled using the program Canu (https://github.com/marbl/canu, v 2.1) [64]. The Canu first built read databases (gkpStore) with the settings “min ReadLength = 1,000” and “corOutCoverage = 40”, then built overlap databases (ovlStore) with the settings “minOverlapLength = 500”, and lastly performed an error correction through falcon_sense method (option “correctedErrorRate = 0.025”). Then, Illumina paired-end reads were also aligned to consensus assembly using BWA and the initial draft assembly was polished twice using the Arrow with the setting “miniCoverage = 15”. Pilon (v 1.23) (default parameters) [65] was used to correct the assembled contigs again. According to the redundancy in the assembly results, we subsequently used Purge_haplotigs (https://bitbucket.org/mroachawri/purge_haplotigs.git) (default parameters) to reduce the redundant sequences and obtained the final assembled contigs. Finally, the assembled contigs were corrected mis-joins, orders, orients, and anchored contigs from the draft assembly into a candidate chromosome-length assembly by Hi-C data using Juicer (v 1.5) (default parameters) [66] and 3D-DNA (v 180922) (default parameters) [67]. To further improve the quality and interactive correction, we reviewed the candidate assembly with JuiceBox Assembly Tools (https://github.com/theaidenlab/juicebox). Last, BUSCO (v 4.1.4) was used to estimate the genome quality.
Genome annotation
Tandem repeats in the loach genome were detected using Tandem Repeats Finder (v 4.09; default parameters) [68]. Repeat elements in the loach genome were annotated using both de novo and homology-based methods. Transport elements (TEs) in the loach genome were identified using RepeatMasker (v 4.0.6) and RepeatProteinMask (v 1.36) with default parameters based on the RepBase database (v 21.12) [69]. For de novo predictions, a de novo transposable element library was constructed using RepeatModeler (v 1.0.5), which was then used to predict repeats with RepeatMasker (v 4.0.6) with default parameters [70].
Protein-coding genes in the loach genome were annotated by combining ab initio predictions, homology-based prediction, and RNA sequencing (RNA-seq)-based methods. For ab initio annotation, Augustus (v 3.2.3) [71] and GENESCAN (v 1.0) [72] were used. For homology-based prediction, the protein sequences of five species (D. rerio, C. auratus, C. carpio, S. grahami, and S. rhinocerous) were downloaded from NCBI and aligned to the loach genome sequences using TBLASTN (e-value less than 1×10−5). For predicting the protein-coding gene models, Genewise (v 2.4.1) was used to analyze all alignments. For RNA-seq-based method, cleaned RNA-seq reads were assembled into transcripts and then were aligned against the assembled genome to link spliced alignments. Then, results from the three methods were integrated by EVidenceModeler (EVM, v 1.1.1) [73]. Functional annotations of these predicted genes were analyzed using the public function databases. InterProscan (v 4.8) was used to screen proteins against a database (Pfam, v 27.0). Moreover, GO (v 20171220), KEGG (v 89.1), NR (https://www.ncbi.nlm.nih.gov/refseq/about/nonredundantproteins/), Swissprot (uniport: release-201906), and TrEMBL (uniport: release-201906) databases were used for gene functional annotations using BLAST software (v 2.6.0) with e-value of 1×10−5.
Phylogenetic analysis
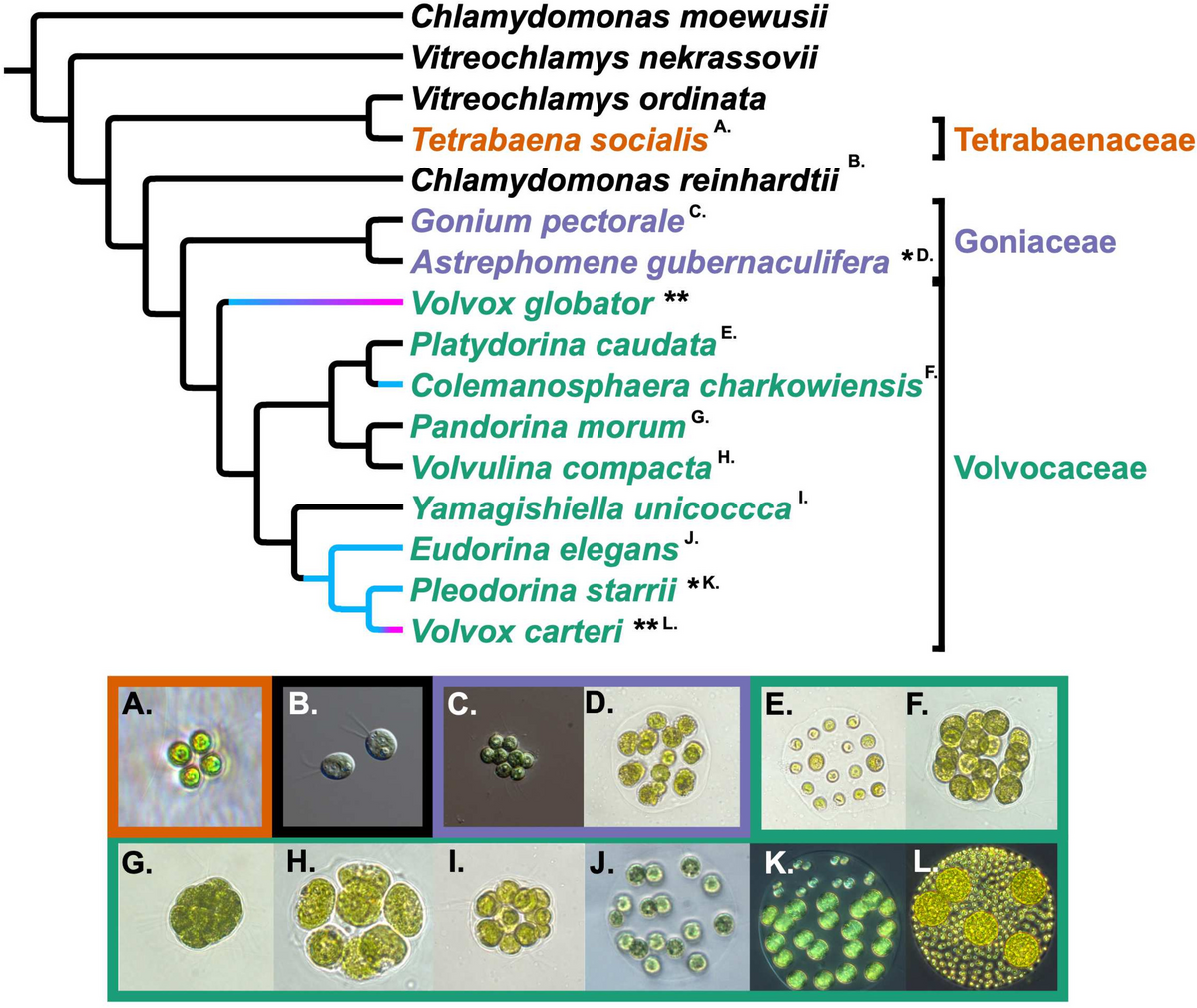
Protein sequences of the loach and other 10 fish species (L. chalumnae, E. calabaricus, L. oculatus, K. marmoratus, O. argus, M. armatus, T. tibetana, D. rerio, C. idellus, and L. rohita) were analyzed by OrthoMCL (v 2.0.9, default parameters) [74] and the protein-coding genes of the 10 species were downloaded from the NCBI database. All the 1818 one-to-one orthologous genes were identified and aligned. Then, the aligned sequences were concatenated into supergenes used for subsequent analyses. The maximum-likelihood method was used to construct the phylogenetic tree using RAxML (v 8.2.10) [75]. The divergence times of these species were estimated through the Bayesian relaxed molecular clock approach using MCMCtree (v 4.8) in PAML package [76]. Fossil records were obtained from TIMETREE website (http://www.timetree.org) and used for calibrating the calculated divergence time.
Gene family expansion and contraction
Based on the results in the previous steps (phylogenetic tree and divergence time analysis), the expansion and contraction of gene families were determined using the CAFE software (v 3.1) with a probabilistic model. The p value for each gene family was calculated, then p value < 0.05 was treated as having a significantly accelerated rate of expansion or contraction. Gene expansion and contraction results for each branch of the phylogenetic tree were estimated, and enrichment analysis about the gene families expanded or contracted in loach was performed with KOBAS (v3.0).
Detection of PSGs
All one-to-one orthologous gene families from the 11 fish species were extracted to identify PSGs. The high-quality multiple-protein alignments were generated and used to estimate three types of ω (the ratio of the rate of non-synonymous substitutions to the rate of synonymous substitutions) using the CodeML program in the PAML package (v 4.8). Branch model (model=2, NSsites=0) was used to detect ω of appointed branch to test (ω0) and average ω of all the other branches (ω1) and the mean of whole branches (ω2). Then χ2 test was used to check whether ω0 was significantly higher than ω1 and ω2 under the threshold p value < 0.05, which hinted that these genes would be under positive selection or fast evolution.
Identification of specific amino acid mutations of fos
Genomes of the loach and other four fish species (O. argus, M. armatus, T. tibetana, and D. rerio) were used for the analysis of fos genes. We collected the genome sequences of O. argus, M. armatus, and T. tibetana from NCBI (http://www.ensembl.org) for comparative analysis. To identify full complement fos genes in the genomes, the Fos protein sequences of zebrafish from ZFIN database (http://zfin.org/) were collected and used as queries to conduct TBLASTN (with e-value of 1×10−5) searches against each of the genomes. Gene annotations for the zebrafish fos gene clusters are shown in Additional file 1: Table S4. GeMoMa2 was used to examine the completeness of the coding sequences. These steps were conducted in a recursive fashion until no new candidates were detected from genome. MAFFT software (default parameters) was used for multiple alignments and then the maximum-likelihood method was used to construct the phylogenetic tree using RAxML (v 8.2.10) [75]. Furthermore, based on the results of fos identification, protein sequences of Fos were aligned using the Clustal W. The domain region of the Fos protein was determined using Pfam (v 1.6).
Whole-mount in situ hybridization (WISH) analysis
To investigate the location of fos (ID: Mis0086400.1) gene in WT loach, a specific Dig-labelling anti-sense RNA probe was synthesized by using T7 in vitro transcriptional polymerase with DIG RNA labelling kit (Roche Molecular Biochemicals, Germany). The probe was amplified from the cDNA pool by using appropriate primers (Additional file 1: Table S5). Healthy WT loaches were selected for reproduction. Loach embryos (n > 60) of 72 hpf and 96 hpf were sampled. All embryos were fixed in 4% (w/v) paraformaldehyde. Then, the WISH of loach embryos was conducted according to a previously described method [77].
Generation of fos (ID: Mis0086400.1) mutant loach
We used a CRISPR-Cas9 strategy to generate a fos mutant loach. The target site of CCAACTTGAGGATGAGAAATCC was determined according to all design principles. Based on our previous study, the Cas9 RNA and gRNA were transcribed in vitro. The construction methods and injection procedures were performed, referring to Sun et al. [78]. The genomic DNA was obtained from tail fin tissues by using Universal Genomic DNA Kit (CWBIO, China) according to the manufacturer’s protocol. A pair of primers that amplified the target genome region was designed for mutation analysis (Additional file 1: Table S5). The amplified DNA fragment was cloned into pMD-19T vectors and then sequenced. The fos mutant ones (F0 generation) were crossed with WT loach to produce F1 generation. Then, the heterozygous F1 generation with the same mutation sequences (fos+/− loach) was self-crossed and the offspring was checked for homozygous mutants (fos−/− loach). Quantitative PCR (qPCR) was used to detect the expression levels of fos in liver tissues of WT and fos−/− loach. The survival rates of loach fertilized eggs were recorded. fos−/− loaches were used for further analysis.
qPCR analysis
Total RNAs of liver tissues (n=9) were extracted by using RNA isoPlus (TaKaRa, Japan). qPCR conditions were as follows: 95 °C for 30 s followed by 40 cycles consisting of 95 °C for 5 s and 57 °C for 30 s. The fluorescent flux was then recorded, and the reaction continued at 72 °C for 6 s and 95 °C for 5 s. Finally, expression levels of target genes were calculated by using the 2−ΔΔCT method. β-actin was used as the reference gene for normalization. All the procedures were based on the methods described by Liu et al. [79]. All of the primer sequences for qPCR are listed in Additional file 1: Table S5.
Micro-computed tomography (Micro-CT) analysis
The whole bodies of WT, fos−/− loach, and C. sinensis (n=3/group) were fixed in 4% (w/v) paraformaldehyde for 48 h. Before Micro-CT scanning, these fish were washed by phosphate buffer solution (PBS), and then scanned using Skyscan high-resolution micro-CT (CT skyscan1276, Bruker, USA). All three-dimensional (3D) images were reconstructed using the CTan1.17 and CTvox program and analyzed using DataViewer software. All fish were imaged at an isotropic voxel size of 13 μm using an X-ray tube potential of 55 kVp, a 0.50-mm aluminum filter, an X-ray intensity of 0.20 mA, and an integration time of 406 ms per slice for vertebrae. Quantitative and qualitative analyses of bone parameters were performed within a square region of interest set at 0.50 mm below the growth plate. The bone morphometric parameters including BMD, BV/TV, and Tb.Th were analyzed.
Tartrate-resistant acid phosphatase (TRAP) staining and toluidine blue staining
The spine tissues from WT (n=6) and fos−/− loach (n=6) were sampled and fixed in 4% paraformaldehyde, dehydrated using a graded alcohol series, and embedded in paraffin. Cross sections of 5-μm thickness were stained with TRAP or toluidine blue. Stained sections were observed under a light microscope (Soptop EX20, China).
Expansion of hemoglobin genes and contraction of digestion/absorption-related genes
Genomes of the loach and other four fish species (O. argus, T. tibetana, D. rerio, and C. idellus) were used for the analysis of hemoglobin (hb) genes. We collected the genome sequences of C. idellus from NCBI (http://www.ensembl.org) for comparative analysis. To identify full complement hb genes in the genomes, the Hb protein sequences of zebrafish were collected according to the method described by Lei et al. [41] and used as queries to conduct TBLASTN (with e-value of 1×10−5) searches against each of the genomes. Gene annotations for the zebrafish hb gene clusters are shown in Additional file 1: Table S4. MAFFT software (default parameters) was used for multiple alignments, and then the maximum-likelihood method was used to construct the phylogenetic tree using RAxML (v 8.2.10). The contracted digestion/absorption-related genes (atp1a1, atp1a2, atp1a3, ryr1, ryr2, and ryr3) in the loach were also identified in the other four fish species genomes, and then we constructed the phylogenetic tree.
Histological observations of posterior intestine tissues
Posterior intestine tissues were sampled from the loach (with intestinal air-breathing) and L. elongata (without air-breathing) (n=6/group), fixed in 4% paraformaldehyde for 24 h, and dehydrated in graded ethanol and embedded in paraffin wax. Cross sections of 5-μm thickness were stained with hematoxylin and eosin (H&E) and prepared for light microscopy, according to the method described by Cao and Wang [80]. Based on the results of H&E staining, we analyzed the difference of posterior intestine tissues between M. anguillicaudatus and L. elongata.
Identification of air-breathing-related genes of the loach
To better identify air-breathing-related genes of the loach, together with our previous studies including developmental transcriptome analysis of loach posterior intestines [42], intestinal air-breathing inhibition trial (transcriptome [10] and microRNAs [43] analysis), and comparative transcriptome analysis of posterior intestines between the loach and non-air-breathing L. elongata [14], an air exposure trial (namely, an intestinal air-breathing enhancement trial) was performed and the procedures in details were described as Sun et al. [12]. A total of 18 WT loaches were used for transcriptome analysis. Among them, nine loaches were randomly selected for posterior intestine sampling, namely, the control group (C_chang). Then, the remaining nine individuals were placed on moist towels (air exposure group (T_chang)). After 6 h air exposure, posterior intestine tissues were sampled from T_chang groups. The samples were stored at 80 °C for RNA isolation. cDNA library of each tissue was prepared and then sequenced on the Illumina sequencing platform by Majorbio, Inc. (Shanghai, China). Gene expression levels were calculated using StringTie (v 2.1.0) with the fragments per kilobase of exon model per million mapped fragments (FPKM) method. The DEGs were identified using DESeq2. Genes with |log2 fold change| ≥ 1 and FDR< 0.05 were considered to be DEGs. GO functional enrichment analysis and KEGG pathway analysis were carried out using Goatools and KOBAS, respectively. Then, the RNA-Seq data was validated by qPCR. In addition, the expression levels of hba and hbb genes in posterior intestines of the loach under air exposure lasted for 12 h, were measured.
The genomes of loach and other four fish species (O. argus, M. armatus, T. tibetana, D. rerio, and C. idellus) were used to analyze the air-breathing-related genes. The detailed steps for the identification of the air-breathing-related genes were the same as the fos gene. Gene annotations for the air-breathing-related gene clusters in zebrafish genome are shown in Additional file 1: Table S4. These genes were identified by TBLASTN (with e-value of 1×10−5). MAFFT software (default parameters) was used for multiple alignments, and then the maximum-likelihood method was used to construct the phylogenetic tree using RAxML (v 8.2.10).
Identification of FMO and UGT gene families
The genomes of the loach and other four fish species (T. tibetana, D. rerio, O. mossambicus, and P. fulvidraco) were used in the analysis of fmo5s and UGT gene family. We collected the genome sequences of O. mossambicus and P. fulvidraco from NCBI (http://www.ensembl.org) for comparative analysis. To identify full complement fmo and ugt genes in the genomes, the Fmo and Ugt protein sequences of zebrafish from ZFIN database (http://zfin.org/) were collected and used as queries to conduct TBLASTN (with e-value of 1×10−5) searches against each of the genomes. Gene annotations for the zebrafish fmo and ugt gene clusters are shown in Additional file 1: Table S4. The detailed steps for the identification of FMO and UGT gene families were the same as the fos gene. MAFFT software (default parameters) was used for multiple alignments, and then the maximum-likelihood method was used to construct the phylogenetic tree using RAxML (v 8.2.10).
Drug stress trial
To scan the genes involved in noxious environmental adaptation of the loach, a drug stress trail (Benzopyrene (LC50=20 μg/L), 1-Naphthol (LC50=8 mg/L), Pyrene (LC50=300 μg/L), p-Nitrophenol (LC50=10 mg/L), and Bisphenol A (LC50=8 mg/L)) was performed, and WT loach adults were used here. The 24 h LC50 of different drugs were determined by our pre-experiment. After the treatment, liver tissues of nine loaches from each group (five drug treatment groups and one control group (without drug treatment)) were sampled and sequenced on the Illumina sequencing platform by Majorbio, Inc. (Shanghai, China). For each group, the liver tissues from three loaches were mixed as a biological sample. The detailed analysis steps of RNA-seq were the same as the above mentioned.
Gene expression analysis during drug stress trail
Based on the RNA-seq results, we performed another drug stress trial. During the trial, the liver tissues (n=9) were sampled at different time points (0, 1, 6, 12, and 24 h) for qPCR and Western blotting (WB) analysis. All of the primer sequences for qPCR are listed in Additional file 1: Table S5. Glyceraldehyde-3-phosphate dehydrogenase (Gapdh) was used as the reference protein.
Expression and location analysis of fmo5 (ID: Mis0185950.1) in the loach
Ten tissues (i.e., fin, blood, muscle, gill, posterior intestine, skin, spleen, barbel, liver, and anterior/mid intestine) were sampled from WT loach adults (n=6). Total RNAs of the tissues were extracted by using MolPure® Cell/Tissue miRNA Kit (Cat No.19331ES08; Yeasen, Shanghai, China) and used for qPCR analysis. All of the primer sequences for qPCR are listed in Additional file 1: Table S5.
The WISH analysis of loach fmo5 was performed here. For immunohistochemical (IHC) analysis, the liver and intestine tissues from WT loach adults (n=6) were sampled and fixed in 4% paraformaldehyde. The procedures of immunohistochemical were performed as described previously with slight changes [81]. The polyclonal antibody of FMO5 (Rabbit, A7673, 1:1000, ABclonal, China) was used as the primary antibody and the peroxidase-conjugated goat anti-rabbit IgG (HRP Goat Anti-Rabbit IgG, ABclonal AS014, 1:10000) was used as the second antibody. Tissues were observed under a light microscope (Soptop EX20, China).
Generation of fmo5 (ID: Mis0185950.1) mutant loach and drug stress trial
The target site of GGATGTAGAGACAGAGTCGAAGG was determined according to all design principles. The detailed steps for fmo5 mutation generation were the same as the fos gene mutant. Moreover, qPCR and WB techniques were used to detect the expression levels of fmo5 in liver tissues of WT (n=6) and fmo5−/− (n=6) loach. The body weights of WT (n=30) and fmo5−/− (n=30) loach were recorded. fmo5−/− loach adults were used for further analysis.
We here conducted another drug stress trial on WT (n=60) and fmo5−/− (n=60) loach, with the same 24 h LC50 of each drug. During the treatment, we recorded the survival rate every 12 h until 96 h.
WB analysis
The liver tissues from WT and fmo5−/− loach were used for WB analysis. The polyclonal antibody of FMO5 and anti-GAPDH (Rabbit, AC001, ABclonal, China) were diluted 1000 and 2000 times with the primary antibody dilution buffer (Gbcbio Technologies Inc., China), respectively. The peroxidase-conjugated goat anti-rabbit IgG (HRP Goat Anti-Rabbit IgG, ABclonal AS014, 1:10000) was selected as the second antibody. Detailed procedures were described by Ida et al. [82].
Alcian blue-periodic acid-Schiff staining (AB-PAS staining) of loach intestines
Sections of the anterior intestine tissues from WT (n=6) and fmo5−/− (n=6) loach were stained by AB-PAS staining (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) according to the manufacturer’s instructions. We then measured the mucosal fold height. The number of goblet cells in each 100 μm mucosal fold height (N for short) was calculated according to the formula: N = (n (the total number of goblet cells)/n (the total number of mucosal folds)) / (h (the average height of mucosal folds)/100 μm)). The average height of mucosal folds was normalized by the length of the corresponding loach.
Histological observations of loach liver tissues
H&E staining and immunofluorescence analysis of liver tissues from WT (n=6) and fmo5−/− (n=6) loach were carried out for histological observations. The procedures of immunofluorescence were performed as described previously with slight modifications [83]. The polyclonal antibody of Chop (produced by our laboratory, Rabbit, 1:50) was used as the primary antibody and Cy3-labeled goat anti-mouse antibody (1:200, ABclonal, China) used as second antibody. The nuclei were stained with DAPI. Tissues were observed under a laser scanning confocal microscope (Leica DMi8, Germany).
Expression analysis of ER stress-related genes in intestine and liver tissues
The intestine and liver tissues were sampled from WT (n=6) loach and fmo5−/− (n=6) loach for qPCR and WB analysis. We investigated the expression profiles of some ER stress-related genes (atf6, eif2ak3, and chop) in liver and intestine tissues and muc2 in intestines at mRNA and protein levels. All of the primer sequences for qPCR are listed in Additional file 1: Table S5. The polyclonal antibodies of Atf6 (Rabbit, 1:1000, A0202, ABclonal, China), Eif2ak3 (Rabbit, 1:1200, A18196, ABclonal, China), Muc2 (Rabbit, 1:1000, A14659, ABclonal, China), and Chop (produced by our laboratory) were diluted 50 times with the primary antibody dilution buffer (Gbcbio Technologies Inc., China).
Statistical analysis
All data were presented as the mean ± standard deviation (SD). Statistical analyses were performed using SPSS 26.0 software (IBM Analytics, Richmond, VA, USA). For two group comparison, a t-test was performed. One-way ANOVA was performed followed by Tukey’s test for multiple comparisons. A p value < 0.05 was considered significant, while p < 0.01 and < 0.001 indicated a very significant difference and an extremely significant difference, respectively.



留言 (0)