
記住我

Trem2 H157Y knock-in mice were generated via CRISPR/Cas9 by the Hope Center Transgenic Vectors Core of the Washington University [25]. CRISPR gRNAs for in vitro testing were identified using CRISPOR (http://crispor.tefor.net/) and synthesized as gBlocks (Integrated DNA Technologies, IDT) with the sequence 5’GGAGGTGCTGTgTTCCACTT3’. In vitro target specific gRNA cleavage activity was validated by transfecting N2A cells with PCR amplified gRNA gblock and Cas9 plasmid DNA (px330, addgene) using ROCHE Xtremegene HP. Cell pools were harvested 48 h later for genomic DNA prep, followed by sanger sequencing of PCR products spanning the gRNA/Cas9 cleavage site, and TIDE analysis (https://tide.nki.nl/) of sequence trace files. CRISPR sgRNA (IDT, 20 ng/ul) and Cas9 (IDT, 50 ng/ul) proteins were complexed to generate the ribonucleoprotein (RNP) for injection along with a 200 nucleotide ssODN donor DNA (synthesized by IDT, 20 ng/ul), 5’tatatcttgtcctttgctgatctgtttgccctgggacctccatcc gcagtcactgccagggggtctaagaagggaccactactgtacCTGGAGGTGCTGTaTTCCACTTGGGCACCCTCGAAACTCGATGACTCCTCGGGGACCCAGAGATCTCCAGCATCTTGGTCATCTAGAGGGTctgtaatagacaaaccatgagg3’. All animal work were approved by institutional IACUC protocols. B6/CBA F1 mice at 3–4 weeks of age (JAX Laboratories, Bar Harbor ME, USA) were superovulated by intraperitoneal injection of 5 IU pregnant mare serum gonadotropin, followed 48 h later by intraperitoneal injection of 5 IU human chorionic gonadotropin (PMS from SIGMA, HGC from Millipore USA). Mouse zygotes were obtained by breeding B6/CBA stud males with superovulated B6/CBA females at a 1:1 ratio. One-cell fertilized embryos were injected into the pronucleus and cytoplasm of each zygote. Microinjections and mouse transgenesis experiments were performed as described previously [26, 27]. Founder genotyping was through deep sequencing (MiSeq, Ilumina). Mosaic founders were crossed to WT to generate heterozygous F1 offspring, which were also deep sequenced to confirm correctly targeted alleles. In addition, we performed off-target analysis with two heterozygous F1 mice from each of the two founders (1 and 2) using the online tool CRISPOR (http://crispor.tefor.net/) [28]. Three putative sites with top CFD scores above 0.3 were identified and examined by Sanger sequencing (GENEWIZ) of PCR amplification products using extracted genomic DNA. No off targets were identified in mice from both founders.
The genotype of Trem2 in all the mice used for experiments was characterized by quantitative PCR (qPCR) with Custom TaqMan SNP Genotyping assays (Thermo Fisher). All the mice were housed in a temperature-controlled environment with a 12-h light–dark cycle and free access to food and water. All animal procedures were approved by the Mayo Clinic Institutional Animal Care and Use Committee (IACUC) and in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals.
Introduction of Trem2 H157Y mutation to 5xFAD amyloid mouse modelTrem2 H157Y homozygous mice (Trem2H157Y/H157Y) were crossed with 5xFAD mice (The Jackson Laboratory, stock # 34,848) to obtain the 5xFAD; Trem2H157Y/+ offspring. 5xFAD; Trem2H157Y/+ mice were used to setup breeding cages to establish the littermate cohorts with three genotypes including 5xFAD; Trem2+/+, 5xFAD; Trem2H157Y/+, and 5xFAD; Trem2H157Y/ H157Y. The genotype of 5xFAD mice was characterized through probe-based qPCR with the protocol provided by the Jackson Laboratory. All the 5xFAD mice used as breeders or in our experimental cohorts were hemizygous.
Hippocampal LTP recordings and analysesElectrophysiological recordings were performed with littermates of Trem2 H157Y homozygous mice and WT mice at 6 months of age as previously described [29] with minor modifications. Each mouse was acutely decapitated and the brain was dissected out to conduct transverse slicing in ice-cold cutting solution containing 110 mM sucrose, 60 mM NaCl, 3 mM KCl, 1.25 mM NaH2PO4, 28 mM NaHCO3, 0.6 mM sodium ascorbate, 5 mM glucose, 7 mM MgCl2 and 0.5 mM CaCl2. Field excitatory post-synaptic potentials (fEPSPs) were obtained from area CA1 stratum radiatum with the use of a glass microelectrode (4-6 mΩ) filled with artificial cerebrospinal fluid (aCSF) containing 125 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 25 mM NaHCO3, 25 mM glucose, 1 mM MgCl2 and 2 mM CaCl2. fEPSPs were evoked through stimulation of the Schaffer collaterals using a 0.1 ms biphasic pulse delivered every 20 s. After a consistent response to a voltage stimulus was established, to obtain the profile for input–output curve (I/O curve), the voltage was increased from 0 mV with a step of 0.5-1 mV for 30 sweeps and the inter-sweep interval is 5 s. For each individual slice, all other stimulation paradigms were induced at the same stimulus voltage which produces the 50–60% of the maximum fEPSP amplitude. Paired-pulse facilitation (PPF) was induced with paired-pulses given with an initial delay of 20 ms and the inter-pulse interval incrementally increased 20 ms until a final delay of 400 ms was reached. The fEPSP baseline responses were then recorded for 20 min. The tetanus used to evoke LTP was a theta-burst stimulation (TBS) protocol consisting of five trains of four pulse bursts at 200 Hz separated by 200 ms, repeated six times with an inter-train interval of 10 s. Following TBS, fEPSPs were recorded for 60 min.
All analyses were performed by customized programming in MATLAB (2021b). The fEPSP slope was calculated within the first 1 ms of the descending domain. I/O curve was presented as the fEPSP slope versus fiber volley amplitude responding to increasing stimulus intensities. PPF strength was examined by the ratio of the second fEPSP slope to the first fEPSP slope in each stimulation pair. Long term potentiation profile was assessed in each minute before and after TBS as the mean fEPSP slope normalized to the mean fEPSP slope of baseline recordings.
Primary microglia cultureCortical cells from pups (p1-p3) were isolated, filtered with 100 um cell strainers (Falcon, 352,360), and plated in T75 flasks (Genesee, 25–209) with high-glucose DMEM medium (Gibco, 11965084) containing 10% Fetal Bovine Serum (FBS). Medium was changed to medium containing 25 ng/mL recombinant mouse GM-CSF (Gemini Bio, 300-308P) the next day. Tails from each pup were kept for genotyping. Five days after cell plating, medium in each flask was replaced with fresh GM-CSF-containing medium. On day 9 or 10, microglia were collected by shaking the flasks at 200–220 rpm at room temperature (RT) for ~ 20 min, resuspended in non-GM-CSF containing medium, and plated into 6-well plates. After 24 h, medium from each well was collected as conditioned medium. Cells were lysed with RIPA buffer (Millipore, 20-188) supplemented with protease inhibitor (cOmplete, Roche) and phosphatase inhibitor (PhosSTOP, Roche) followed by mild agitation at 4 °C for 30 min and centrifugation at 20,000 g at 4 °C for 30 min. Supernatant was collected as RIPA lysate.
Microglia isolation from adult miceMicroglia were isolated from adult mouse brains as previously described [30]. In brief, mice were transcardinally perfused with 0.01 M PBS followed by cortex dissection from both hemispheres. The cortices were dissociated with papain-based enzyme mix (Miltenyi, 130–092-628) using gentleMACS™ Dissociator (Miltenyi, 130-093-235) followed by debris removal through filtering with the 70-µm cell strainer. Myelin was then removed through magnetic sorting after the incubation with myelin removal beads (Miltenyi, 130-096-731). The obtained single cell suspensions were incubated with CD11B-conjugated beads (Miltenyi, 130-049-601) followed by magnetic sorting for CD11B+ microglia. Around 300 K microglia were captured per sample and lysed with RIPA buffer supplemented with the protease and phosphatase inhibitors followed by mild agitation at 4 °C for 30 min and centrifugation at 20,000 g at 4 °C for 30 min. Supernatant was collected as RIPA lysate and subject to Western blotting.
Tissue preparation for immunofluorescence staining, biochemical assaysBlood samples were collected from mice vena cava after isoflurane induced deep anesthesia, stored at 4 °C overnight and subsequently centrifuged at 1000 g for 10 min to collect the supernatant as serum. After blood collection, mice were transcardinally perfused with 0.01 M PBS and the brains were dissected out. Half of the brain was fixed in 4% paraformaldehyde (PFA, Fisher Scientific) for 24 h, dehydrated with 30% sucrose (Sigma) for 48 h, embedded in O.C.T. compound (SAKURA) and snap-frozen in liquid nitrogen before cryostat sectioning. The other hemisphere was dissected into cortex, hippocampus, midbrain, and cerebellum which were snap-frozen in liquid nitrogen and stored at -80 °C. The cortices were then pulverized (CP02 cryoPREP Automated Dry Pulverizer) and divided into two aliquots: 20–30 mg for RNA extraction and 55–65 mg for protein extraction.
Cortical proteins were extracted sequentially with different lysis buffers. Cortical powder was homogenized in Tris-buffered saline (TBS, Fisher Bioreagents, BP2471-500, 600 µl) supplemented with protease inhibitor (cOmplete, Roche) and phosphatase inhibitor (PhosSTOP, Roche) and subjected to ultracentrifugation at 100,000 g at 4 °C for 1 h. The supernatant was collected as TBS lysate with a protein concentration ~ 3 µg/µl. The pellets were then resuspended in TBSX (TBS plus 1% Triton-X100, 600 µl) supplemented with protease inhibitor and phosphatase inhibitor, homogenized, and mild agitated at 4 °C for 30 min followed by ultracentrifugation at 100,000 g at 4 °C for 1 h. Supernatant was collected as TBSX lysate with a protein concentration ~ 3.5 µg/µl for non-amyloid bearing mice and ~ 5 µg/µl for amyloid bearing mice. For amyloid bearing mice, the pellet was further resuspended in 5 M guanidine hydrochloride (GND, Sigma, 600 µl) followed by sonication and centrifuged at 100,000 g for 1 h at 4 °C. The supernatant was collected as GND lysate with a protein concentration ~ 1.6 µg/µl. Total protein concentration in each lysate was measured (Pierce™ BCA Protein Assay Kit, Cat# 23225) before transferring to 96-well storage plates or 1.5 ml tubes and stored at -80 °C until further analysis.
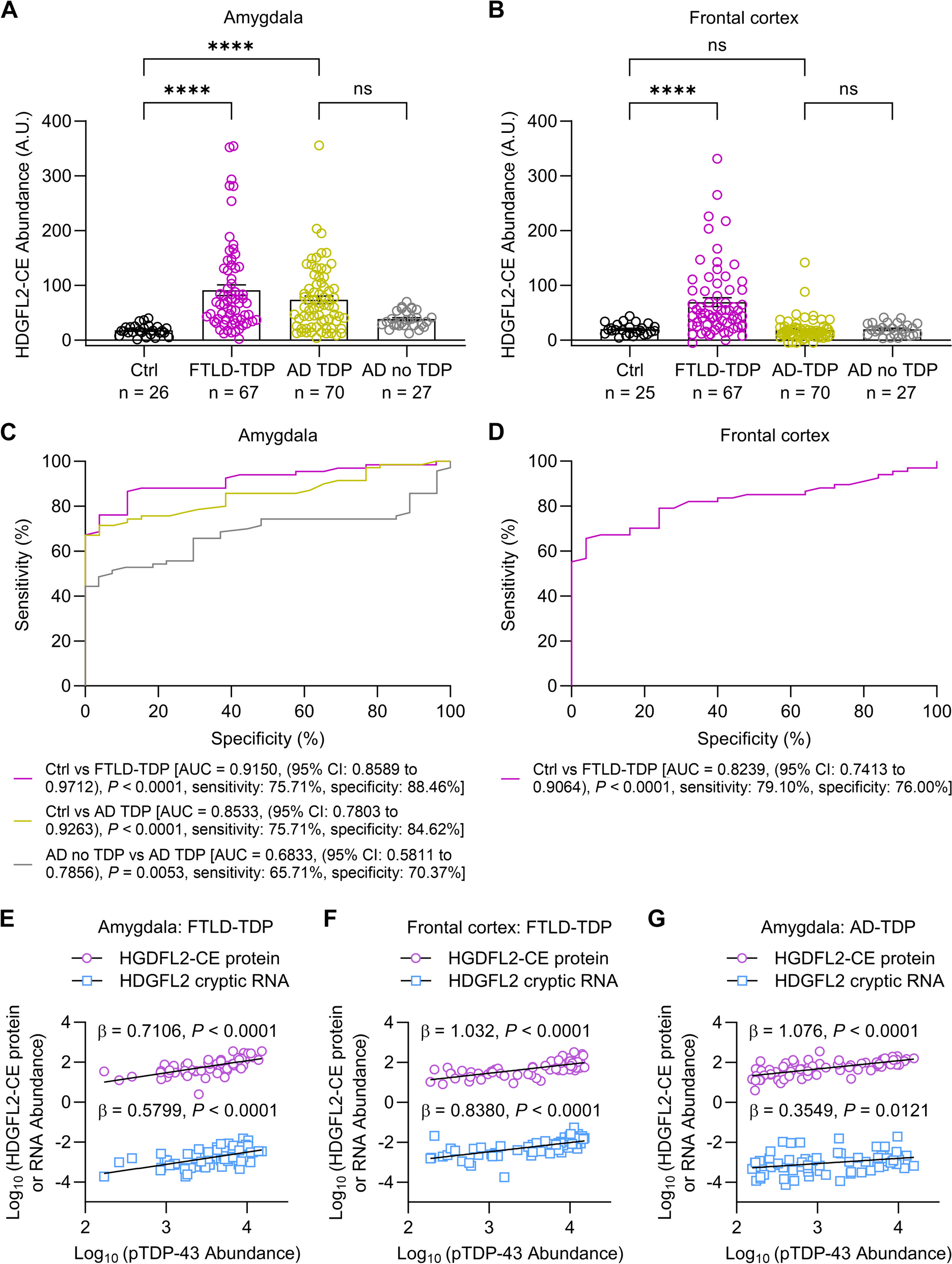
Immunofluorescence staining, X34 stain and quantificationThe embedded hemispheres were coronally sectioned at a 40 µm thickness. Referencing the mouse brain atlas (Paxinos & Franklin, 2013), sections located from AP -1.7 mm to AP -2.06 mm were selected for the following procedures. First, brain slices were blocked in blocking buffer (5% goat serum plus 0.25% Triton in PBS) for 1 h at RT, then incubated overnight in primary antibody solution at 4 °C. Slices were then incubated in the Alexa Fluor-conjugated secondary antibodies solution (1:1000, Invitrogen) at RT for 2 h. We used the following antibodies against IBA1 (Wako, 019-19,741, 1:1000), Aβ (MOAB2, Abcam, ab126649, 1:1000), CD68 (Bio-Rad, MCA1957,1:500), LAMP1 (Abcam, ab25245, 1:500), APP (Millipore Sigma, MAB348, 1:300), and GFAP (Millipore Sigma, MAB360, 1:500). To detect TREM2, 5% BSA and 0.25% Triton X-100 in PBS was used for blocking and preparation of TREM2 antibody solution (R&D, AF1729, 1:300). Fibrillar Aβ plaque staining used free-floating sections from 5xFAD mouse cohorts. Sections were permeabilized with 0.25% Triton X-100 in PBS and stained with 10 µM X-34 (Sigma, SML1953) in a mixture of 40% ethanol and 0.02 M NaOH in PBS [31]. Following X34 stain, fluorescence immunostaining targeting proteins of interest were performed on the same slice.
To quantify signals of Aβ, X34, IBA1, LAMP1, APP, IBA1, CD68, GFAP and TREM2, images were taken, and stitched using Keyence (BZ-X800) at 20X. For each staining, the hippocampus and the cortex region located above the hippocampus were traced and saved as region of interest (ROI) images in Image J. A unified intensity threshold was applied to all the sample images in each staining. Pixels with the signal intensity above the threshold were used to calculate area percentages of positive immunoreactive signals in ROI through the particle analysis plugin. For Aβ and X34 staining, plaque numbers and sizes were also assessed. Plaque densities were calculated using plaque (diameter > 8 µm [32, 33]) numbers divided by the ROI areas. For IBA1 staining in the non-amyloid mice, a unified intensity threshold was applied to recognize the microglial cell body followed by particle analysis to examine the microglial density and cell body size. To further assess microglial morphology, 4–5 fields were taken per sample under confocal (Zeiss) at 20 × with a zoom factor 0.6. Images were processed to remove background and skeletonized followed by analysis of branch number, junction number and total branch length per microglia [34].
To assess the interaction of microglia and plaque, we co-stained X34 and IBA1. 30-40 z-stack images per sample were taken under Confocal (Zeiss) at 40X with a zoom factor 0.6. Plaque-centered ROI was traced with a radius of 30 µm and saved through Image J. The number of microglia surrounding each plaque within the radius of 30 µm were manually counted. Colocalization of IBA1 and X34 was decided through ‘Colocalization Threshold’ analysis in Image J.
All the analyses were conducted in a batch mode through customized macro coding in Image J and MATLAB (2021b) with the same setting parameters for all the samples. Researchers were blinded to genotypes and groups when performing and quantifying the immunofluorescence staining.
Aβ40, Aβ42, Aβ oligomer, sAPPα, sAPPβ, CTFβ, TNFα, and TREM2 ELISAAβ40 and Aβ42 levels in TBS, TBSX, and GND lysates were determined by ELISA as previously described [35] using an end-specific Aβ monoclonal antibody (13.1.1 for Aβ40 and 2.1.3 for Aβ42) and an HRP-conjugated detection antibody (in-house Ab5 antibody) [36]. Aβ42 in ISF was detected by commercial kits (Thermo Fisher, KHB3544). Aβ42 oligomers in TBS and TBSX lysates were detected by commercial kits (Biosensis, BEK-2215-2P). Soluble APPα (sAPPα), sAPPβ in TBS lysates were detected by commercial kits (Meso Scale Discovery, K15120E-2). CTFβ in TBSX lysates was detected by commercial kit (IBL, 27776). TNFα was measured in TBS lysates using commercial kit (Meso Scale Discovery, K152QWD-1).
TREM2 in cortical TBS, TBSX lysates, conditioned medium and cell lysates of primary microglia, serum were measured as described [30, 37] with minor modification using the Meso Scale Discovery (MSD) platform. Streptavidin-coated 96-well plates (MSD, L55SA) were blocked overnight at 4 °C in blocking buffer (3% bovine serum albumin and 0.05% Tween-20 in PBS). On the second day, capture antibody (R&D Systems, BAF1729, 0.25 ug/ml) was applied for an incubation at RT for 1 h. After washing with PBST (0.05% Tween-20 in PBS), samples were incubated overnight at 4 °C with an established dilution in fresh-prepared sample buffer (1% bovine serum albumin and 0.05% Tween-20 in PBS) supplemented with protease inhibitor (cOmplete, Roche). Following another wash with PBST, detection antibody (R&D Systems, MAB1729,) was applied for an incubation at RT for 1 h. Sulfo-tag labeled anti rat antibody (MSD, R32AH-5) was applied at RT for 1 h, and final measurements were made with Read Buffer (MSD, R92TC-3). TBS lysate, TBSX lysate, and serum from Trem2-KO mice were used as negative controls.
Parallel reaction monitoring-based targeted quantitation of TREM2 in mouse brainMouse brain tissues were snap-frozen immediately after collection. Brain tissues (2 mg) were lysed in lysis buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1% n-Octylglucoside, HALT protease/phosphatase inhibitor cocktail) by sonication. After centrifugation, the supernatant was transferred as brain lysate and subjected to the following procedures. The protein concentration from the brain lysate was determined by BCA assay. Biotinylated anti-mouse TREM2 antibody (R&D, BAF1729) was bound to Streptavidin-Dynabeads (Life technologies, 29200) by mixing for 30 min at RT. After washing to remove unbound antibody, equal amounts of protein from each sample were incubated with biotinylated anti-mouse TREM2 antibody (R&D, BAF1729) bound to Streptavidin-Dynabeads overnight at 4 °C. The automated KingFish Flex System (ThermoFisher Scientific) was used to wash beads with ice-cold PBS and elute of TREM2 with 5% acetic acid with high reproducibility. The eluents containing TREM2 were subjected to in-solution trypsin digestion. The resulting peptides were spiked with 5 femtomoles (fmol) of stable isotope-labeled internal standard (SIL-IS) peptides unique to sTREM2-WT, flTREM2-WT, sTREM2-H157Y, and flTREM2-H157Y (synthesized by the Mayo Clinic Proteomics Core). Parallel reaction monitoring (PRM) analysis was performed on an Orbitrap Exploris 480 mass spectrometer coupled to Ultimate 3000RSLC NANO LC System (ThermoFisher). The acquired mass spectra were analyzed using Skyline [38,39,40] to calculate the absolute molar amount of sTREM2 and flTREM2 using the relative ratio of native peptides to SIL-IS peptides.
Western blottingEqual amounts of protein from the brain TBS and TBSX lysates, or RIPA lysates from isolated microglia were resolved by SDS-PAGE and transferred to PVDF membranes. After blocking, proteins of interest were detected with appropriate primary antibodies. The membrane was then probed with HRP-conjugated or LI-COR secondary antibodies and visualized using the films or Odyssey infrared imaging system (LI-COR). We used the following primary antibodies against: TREM2 (5F4, ordered from Dr. Haass lab), GLUR2 (Millipore, MAB397, 1:1000), PSD95 (Cell Signaling, 3450 s, 1:1000), synaptophysin (Biolegend, 807,801, 1:1000), SYK (Cell Signaling, 2712S, 1:1000), pSYK (Cell Signaling, 2710S, 1:1000) and β-actin (Sigma, A2228, 1:2000).
RNA extraction, library preparation and sequencingRNA from pulverized cortex was extracted and purified according to our previous study [41]. The RNA integrity numbers (RIN) of all the 40 RNA sequencing (RNAseq) samples were above 9.5. Thus, they were all used for library preparation and sequencing. RNA libraries were prepared from 200 ng of total RNA using the TruSeq RNA Sample Prep Kit (Illumina) according to the manufacturer’s instructions, employing poly-A mRNA enrichment using oligo-dT magnetic beads. The final adaptor-modified cDNA fragments were enriched by 15 cycles of PCR using Illumina TruSeq PCR primers. The concentration and size distribution of the completed libraries were determined using an Agilent Tape Station, (Agilent) and Qubit fluorometer (Invitrogen). Libraries from all the 40 samples were sequenced on Illumina’s NovaSeq 6000 at 75 million fragment reads/sample following Illumina’s standard protocol on a S2 flow cell. S2 flow cells were sequenced at 100 × 2 paired end reads using a NovaSeq S2 sequencing kit, NovaSeq Control Software v1.7.5 and base-calling was analyzed using Illumina’s RTA version 3.4.4.
RNAseq data analysisRNA quantification, quality control and normalizationRNA sequencing reads were processed through the Mayo Clinic RNA sequencing analytic pipeline, MAP-RSeq Version 2.1.1 [42]. Briefly, reads were aligned to the mouse reference genome mm10 using TopHat version 2.1.0 [43] and Bowtie version 1.1.2 [44]. Quality control (QC) was performed using RSeQC version 2.6.2 [45]. Gene counts were generated using featureCounts version 1.4.6-p5 [46]. One Trem2 H157Y (5xFAD) homozygous female sample was excluded from further analysis due to low gene count percentage (39%) and strand-ness check (0.5009). Genes were filtered out from further analyses if there were not at least four samples with 10 counts of the gene. Trimmed Mean of M-values (TMM) normalization was performed with calcNormFactors from the edgeR R package [47].
Differential gene expression, hierarchical clustering and pathway analysisDifferential gene expression analyses were performed in the comparison of Hom vs WT (separately for non-amyloid or amyloid cohorts) using the edgeR quasi-likelihood pipeline [47]. Differentially expressed genes (DEG) were defined with false discovery rate (FDR) < 0.05 and log2|fold change (FC)|> 0.25. Hierarchical clustering was performed in MATLAB using the Clustergram function based on standardized Euclidean distance metric. Volcano plots were generated in MATLAB using –log10 (FDR) as y axis and ± log2 (|FC|) as x axis. Pathway analyses of differentially expressed genes were performed through Ingenuity Pathway Analysis (QIAGEN Inc., https://digitalinsights.qiagen.com/products/ingenuity-pathwayanalysis) [48].
Weighted gene co-expression network analysis and module preservation analysis of the amyloid modules in non-amyloid networkWeighted gene co-expression network analysis (WGCNA) was conducted in non-amyloid mice and amyloid mice, respectively, using residual expression values calculated from adjusting for sex, strand, and exonic rate. Based on the relationship between power and scale independence, the power of 12 was chosen to build scale-free topology using signed hybrid network. We set the minimum modules size as 40 and merged modules whose correlation coefficients were greater than 0.6 (mergeCutHeight = 0.4). Each module was summarized by the first principal component of the scaled module expression profiles, termed module eigengene (ME). For each module, the module membership (MM) was defined as the correlation between gene expression values and ME. Intramodular hub genes are genes with the highest connectivity to other genes within a given module, and were selected based on the p values of MM. To assess the correlation of modules to genotype, we defined the WT genotype as 0 and Hom as 1. Modules were annotated using R package anRichment. MEs of selected modules were compared between genotypes through Wilcoxon rank-sum test. Gene–gene connections among top hub genes were visualized using Cytoscape. GO term enrichment analysis was conducted with anRichment R package.
The preservation of the modules in amyloid network was tested in the non-amyloid network. Separate module preservation analyses were performed for the two datasets using WGCNA. In all analyses, module definitions from the mouse network were used as reference to calculate the z-summary statistics for each module. Z summary score > 2 suggests moderate preservation and Z summary score > 10 suggests strong preservation.
Quantitative PCR (qPCR)Purified RNA (1 µg) was used to prepare cDNA iScript™ Reverse Transcription Supermix (Biorad) and qPCR was performed using the QuantStudioTM 7 Flex Real-Time PCR System (ThermoFisher Scientific). To assess the total Trem2 mRNA, predesigned primers (IDT) targeting exon 4–5 (Mm.PT.58.45957937.g), and customized primers targeting exon 2 (Forward 5′-GCCCATGCCAGCGTGTGGT-3′ and Reverse 5′-CACTGGTAGAGGCCCGC-3′) were used, respectively. Predesigned primers from IDT were used to quantify the mRNA levels of Tyrobp (Mm.PT.58.6069426), Tmem119 (Mm.PT.58.6766267), Cx3cr1 (Mm.PT.58.17555544), C1qa (Mm.PT.58.5375735). The relative gene expression was normalized to Gapdh (IDT, Mm.PT.39a.1) and assessed using the 2−ΔCT method.
In vivo microdialysisTo assess the Aβ clearance, we examined the Aβ level in hippocampal interstitial fluid (ISF) obtained through in vivo microdialysis in awake, free-moving mice as previously described [30, 49, 50]. Animals were placed in a stereotaxic device equipped with dual manipulator arms and an isoflurane anesthetic mask (David Kopf Instruments). Under isoflurane volatile anesthetic, guide cannula (BR style; Bioanalytical Systems) were cemented into the hippocampus (3.1 mm behind bregma, 2.5 mm lateral to midline, and 1.2 mm below dura at a 12˚ angle). Four to six hours post-surgery, a microdialysis probe (30-kilodalton MWCO membrane, Bioanalytical Systems) was inserted through the guide cannula into the brain. Artificial cerebrospinal fluid (aCSF) (mM: 1.3 CaCl2, 1.2 MgSO4, 3 KCl, 0.4 KH2PO4, 25 NaHCO3, and 122 NaCl, pH 7.4) containing 3% bovine serum albumin (BSA; Sigma) filtered through a 0.1 mm membrane was used as microdialysis perfusion buffer. Flow rate was a constant 1.0 ml/min. Samples were collected hourly into a refrigerated fraction collector. The baseline samples were collected for 10 h followed by subcutaneous administration of a \(\gamma\)-secretase inhibitor, LY411575 (5 mg/kg) to rapidly block the production of Aβ. Samples were collected for another 4 h after treatment. ISF Aβ42 in the 14 samples for each mouse was measured by ELISA (Invitrogen, KHB3441, 1:4). To determine Aβ42 half-life [49], datapoints from drug delivery were analyzed. Meeting with the first-order processes, the elimination rate (\(Ke\)) of Aβ42 is related to the slope (\(a\)) of the semi-log plot of concentration versus time: \(a=-Ke/2.3\). The half-life (T1/2) of Aβ42 is further calculated as T1/2\(=0.693/Ke\).
Statistical analysesAll data were reported as mean values ± SEM. Generally, if sample sizes are larger than 7, to ensure that results were valid in the presence of non-normal distributions, or differing variances between groups, Kruskal–Wallis tests with uncorrected Dun’s multiple comparisons or Wilcoxon Rank-sum tests were used. If the sample size ≤ 7 and dataset showed similar variances examined by F-test, unpaired t test was used since nonparametric tests would have very low power. Specifically, One-Way ANOCOVA with comparison of slopes was used in Fig. 2A. Unpaired t test with Welch’s correction (Welch’s t test) was used Fig. 3Q because of the significantly different variances. Wilcoxon matched-pairs signed rank test was applied to Fig. 6B. All the statistical analyses were conducted using GraphPad Prism v8.4.3 or MATLAB. The statistical tests used for each analysis, the sample size and the significance levels were reported in the legend of each figure.
留言 (0)