
記住我






Atypical hemolytic uremic syndrome (aHUS) is an ultra-rare kidney disease characterized by microangiopathic hemolytic anemia, thrombocytopenia, and renal impairment (1). Primary aHUS is associated with genetic and acquired defects that led to dysregulation of the alternative pathway (AP) of complement system, resulting in endothelial damage in the microcirculation of the kidney and other organs (2). About 50% of patients carry genetic abnormalities that affect genes coding for complement regulators (CFH, CD46, CFI and THBD) and components (C3 and CFB), while in 10% of patients anti-FH autoantibodies have been reported (3).
Atypical HUS can be secondary to autoimmune or systemic disease, pregnancy/postpartum, malignant hypertension, drug treatments, cancer and transplantation (4, 5). In the secondary forms, the prevalence of genetic defects is variable, ranging from almost 60% in cases associated with malignant hypertension or pregnancy to less than 10% in drug-induced TMA.
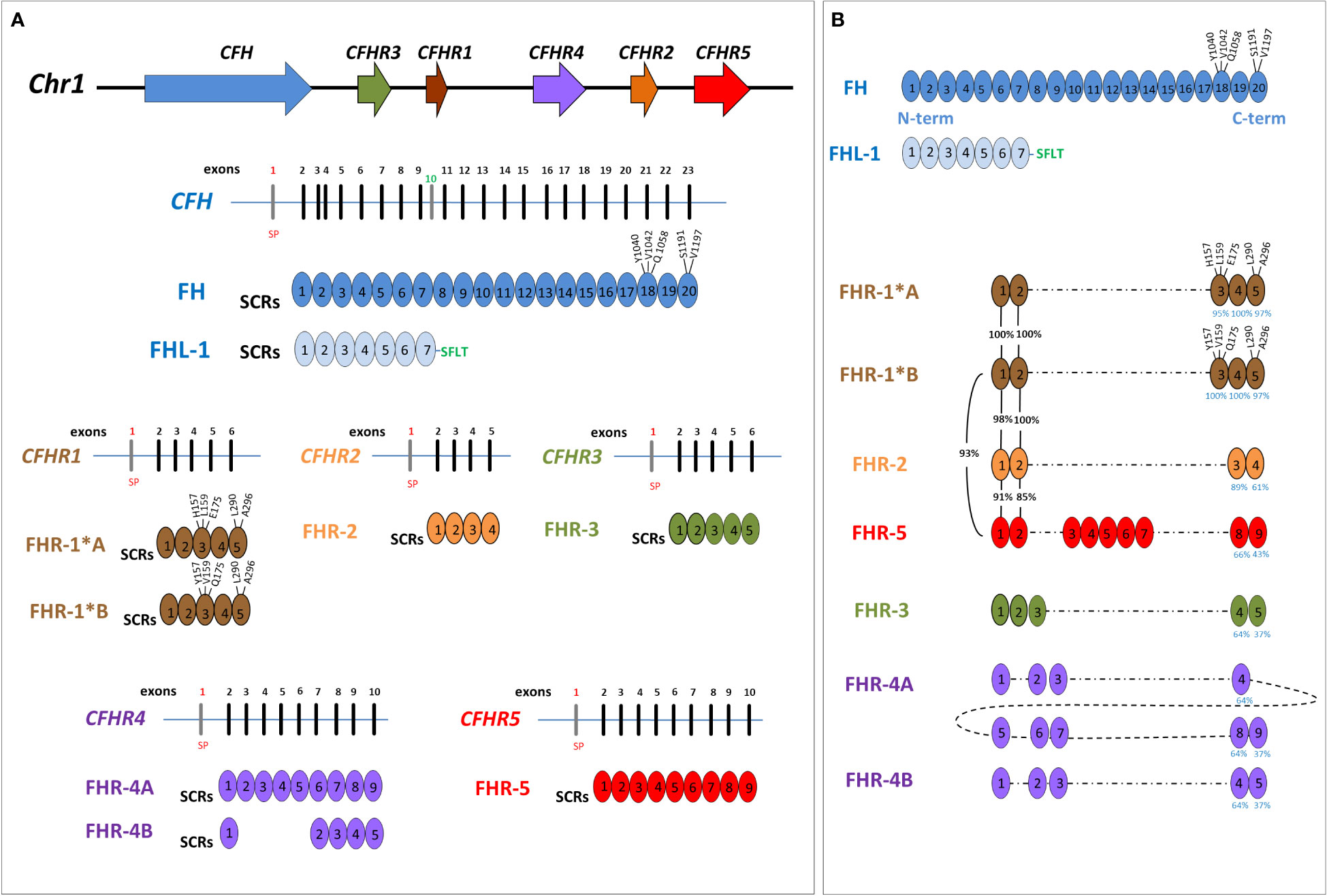
The gene most commonly involved in aHUS is CFH, encoding complement factor H (FH). CFH is mapped on chromosome 1q31 within the RCA (Regulation of Complement Activation) gene cluster, which also includes the CFHR3, CFHR1, CFHR4, CFHR2 and CFHR5 genes, derived from genomic duplication events (6). The resulting FH and FHR proteins are organized in short consensus repeats (SCRs), each consisting of about 60 amino acids (Figure 1), and are mainly produced by the liver and circulate in the blood.
Figure 1 Structure of Factor H family: genes and proteins. (A) The human complement factor H (CFH) gene family is located on chromosome 1q31.3 and includes six genes: CFH, CFHR3, CFHR1, CFHR4, CFHR2 and CFHR5. For each gene, the corresponding protein was represented. Each short consensus repeat (SCR) is composed of about 60 amino acids and is encoded by a single exon, with the exception of SCR2 of Factor H (FH), encoded by exon 3 and 4. Exon 1 of each gene encodes 18 amino acids of the signal peptide (SP). CFH gene is composed of 23 exons and, through two alternative splicing, produces FH, deriving from 22 exons, and Factor H-like protein 1 (FHL-1), deriving from 10 exons. Exon 10 is not included in the FH transcript and encodes the C-terminal four amino acids (Ser-Phe-Leu-Thr; indicated in the Figure with the green SFLT) and the 3’UTR of FHL-1. FHR-1 exists in two isoforms that differ in three amino acids in the SCR3: FHR-1*A is known as acidic isoform and has His at position 157 (H157), Leu at 159 (L159) and Glu at 175 (E175); FHR-1*B is the basic isoform with Tyr at position 157 (Y157), Val at 159 (V159) and Gln at 175 (Q175). (B) Factor H-related proteins (FHRs) share a high degree of conservation within the C-terminal domains of FH (SCR18-SCR19-SCR20), and FHR-1 is the most similar (the percentage of amino acids identity between each SCR of FHR and those of FH is indicated by blue numbers under the SCRs). As represented in the Figure, SCR3 of FHR-1*A differs from SCR18 of FH for 3 amino acids (H157, L159 and E175) while SCR5 differs from FH-SCR20 for 2 amino acids (L290 and A296). At variance, SCR3 of FHR-1*B has the same amino acids of SCR18 of FH. Of note, the N-terminal domains of FHR-1, FHR-2 and FHR-5 (SCR1 and SCR2) have a high sequence identity (indicated by black percentage numbers) and include a dimerization motif which explains their presence in plasma as either homo-or heterodimers. This image was inspired by Jozsi et al. (7) Trends immunology, 2015.
Factor H is the main plasma regulator of the AP of complement and consists of 20 SCRs. FH regulatory activity is mediated by its N-terminal domains (SCR1-4) acting as a cofactor for complement protease Factor I (FI) and accelerating the decay of C3 convertase. In addition, through the SCR6-7 and C-terminal domains (SCR19-20), FH binds C3b and polyanions, such as glycosaminoglycans, heparan sulfate, and sialic acids, and mediates cell surface protection from complement activation. The large majority of CFH genetic abnormalities in aHUS cluster in the C-terminal part of the protein, leading to reduced complement regulation on endothelial cells. However, not all the carriers of heterozygous CFH genetic defects manifest aHUS, due to incomplete penetrance.
FHRs were originally thought to be negative complement inhibitors, but later studies indicated that these molecules may instead enhance complement activation (8). The C-terminal domains of FHRs have a high level of amino acidic sequence identity with SCR18-19-20 of FH leading them to be able to bind the same FH ligands (Figure 1) (8–11). The N-terminal SCRs (SCR1-2) of FHR-1, FHR-2 and FHR-5, are very similar (85-100%) and include a dimerization domain (Figure 1), which explains their presence in the circulation as homo- and hetero- dimers or tetramers (12–14). This oligomerization increases FHR avidity for C3b and C3-opsonized surfaces and for polyanionic surface ligands, which results in the activation of the alternative pathway (12, 13). The N-terminal domains of FHR-3 and FHR-4 (SCR1-3 of both) share high residue sequence similarity with FH SCR6 to SCR8 and FH SCR6-8-9, respectively, which are involved in binding to heparin, C-reactive protein, and microbial surface ligands. However, none of the FHR protein domains have any similarity to the N-terminal regulatory domains of FH, indicating that FHRs lack direct complement regulatory activity, although this point remains controversial (15).
The FH gene cluster is characterized by large repeated regions, which favors genomic rearrangements and copy number variations (CNVs) like the duplications, deletions and inversions that have been reported in association with aHUS and other complement-mediated diseases (16, 17). Genomic alterations involving DNA segments larger than 1kb are defined as structural variants (SVs) and the most frequent SV in the CFH gene cluster is the ~84 kb deletion of CFHR3 and CFHR1 (CFHR3-CFHR1 del), which is associated with a high risk of developing anti-FH autoantibody (anti-FHs)-mediated aHUS (18). Rare SVs involving CFH and CFHRs that lead to hybrid genes such as CFH::CFHR1, CFH::CFHR3, and the reverse CFHR1::CFH hybrids have been reported in patients with primary aHUS, and a few functional analyses have confirmed their involvement in disease pathogenesis (7, 16, 17).
However, data on the prevalence of CFH-CFHR genomic rearrangements in primary and secondary aHUS, their impact on disease penetrance, disease onset, response to therapy and outcome are limited to case reports or case-series.
Here, we report a retrospective study of CFH-CFHR copy number variations (CNVs) in a large cohort of unrelated patients affected by primary (n=258) or secondary aHUS (n=92). We evaluated the prognosis of patients carrying CFH-CFHR SVs and the contribution of the concomitant presence of rare complement gene variants or anti-FHs abnormalities to disease development. To overcome the limits of next generation sequencing (NGS) to detect SVs in the CFH-CFHR region, we applied Multiplex Ligation-dependent Probe Amplification (MLPA), long-read sequencing (Single -Molecule Real-Time, SMRT) and direct sequencing to identify and characterize rare genomic rearrangements. We found them in the 6% of patients, including 2 patients with secondary forms. Furthermore, we identified a group of patients carrying rearrangements that included only CFHR genes, which have so far been reported in association with C3G or Immune complex-mediated membranoproliferative glomerulonephritis (IC-MPGN) (17). This group presented a milder disease phenotype than patients with rearrangements involving CFH.
Our results confirm the important role of CFH genomic rearrangements in the pathogenesis of aHUS and highlight the potential impact of SVs involving CFHR genes in disease predisposition and phenotype.
Material and methodsStudy participantsPatients included in this study were recruited through the International Registry of HUS/TTP, under the coordination of the Aldo and Cele Daccò Clinical Research Center for Rare Diseases (Ranica, Bergamo, Italy).
Clinical information and demographic/laboratory data for patients and their available relatives were collected using a case report form. Biochemical and genetic tests were performed using blood, plasma or serum samples, and DNA was collected for each patient and available relatives.
Healthy controls were recruited among blood donors and were analyzed for copy number variations (CNVs). The samples used for the research were stored at the Centro Risorse Biologiche (CRB) Mario Negri, Malattie Rare e Malattie Renali biobank.
Atypical HUS was diagnosed in all cases with microangiopathic hemolytic anemia and thrombocytopenia (hematocrit less than 30%, hemoglobin level less than 10 g/dL, serum lactate dehydrogenase level higher than 500 U/L, undetectable haptoglobin, fragmented erythrocytes in peripheral blood smear, and platelet count less than 150x103/µl) associated with acute renal failure (serum creatinine>1.3 mg/dl for adults, >0.5 mg/dl for children under 5 years of age and >0.8 mg/dl for children aged 5-10 years old; and/or urinary protein/creatinine ratio >200 mg/g; or an increase of serum creatinine or urinary protein/creatinine ratio>15% compared to baseline levels). Thrombotic thrombocytopenic purpura was ruled out in the presence of ADAMTS13 activity >10% and no anti-ADAMTS13 antibodies. Patients were classified as having primary aHUS when both secondary underlying conditions and Stx-E.Coli infections were ruled out; a secondary form was considered when aHUS was associated with hypertension, autoimmune diseases, infections, pre-existing nephropathy, transplantation, drug exposure or other coexisting conditions (pneumococcal infections and malignancy).
Familial aHUS was diagnosed when two or more members of the same family were affected by the disease at least 6 months apart and exposure to a common trigger infectious agent was excluded. Sporadic aHUS was diagnosed when one or more episodes of the disease manifested in a subject with no familial history of the disease.
The study was approved by the Ethics Committee of the Azienda Sanitaria Locale, Bergamo (Italy) and informed consent was obtained in accordance with the Declaration of Helsinki.
Complement profile assessmentFH and anti-FH autoantibody serum levels were measured using Enzyme-Linked Immunosorbent Assay (ELISA) as previously reported (3).
Genetic screening and biochemical testingGenomic DNA was extracted from peripheral blood leukocytes (Nucleon™ BACC2 kit, GE Healthcare; NucleoSpin Blood columns, Macherey-Nagel). All coding exons and the intronic flanking regions of membrane cofactor protein (CD46), complement factor H (CFH), complement factor I (CFI), complement factor B (CFB), complement C3 (C3) and thrombomodulin (THBD) genes were amplified by polymerase chain reaction (PCR) and were directly sequenced (48-capillary 3730 DNA Analyzer), as previously reported (19). Patients recruited more recently were analyzed using a next generation sequencing (NGS) panel for the simultaneous sequencing of CFH, CD46, CFI, CFB, C3, and THDB through Ion Torrent platform (Life technologies). Since recessive LPVs in the gene encoding diacylglycerol kinase DGKε (DGKE) have been identified in patients with aHUS with an onset in infancy, we also sequenced DGKE, by NGS, in patients carrying uncommon SVs and with a disease onset below 4 years (20, 21).
Patients with uncommon SVs and their available relatives, were genotyped by NGS or direct sequencing for the CFH and CD46 single-nucleotide polymorphisms (SNPs) that define the aHUS-risk haplotypes CFH-H3 and CD46GGAAC, respectively (19, 22–25).
Genetic variants with a reported minor allelic frequency (MAF) below 0.001 in the Genome Aggregation Database (gnomAD) and with a Combined Annotation Dependent Depletion (CADD) phred score ≥10 were considered likely pathogenic variants (LPVs).
CFH-CFHR copy number variationsMultiplex ligation dependent probe amplification (SALSA MLPA P236, MRC Holland, Netherlands) and in-house probes for CFHR4 and CFHR5 were used to evaluate the presence of copy number variations (CNVs) in CFH, CFHR1, CFHR2, CFHR3, CFHR4, and CFHR5 genes in all the patients, as previously reported (17).
Two hundred and fourteen healthy subjects were also analyzed for CFHR4 CNVs using multiplex polymerase chain reaction (mPCR) amplifying intron 1 and exon 2 of CFHR4 and intron 3 of CFHR1 (18).
Single molecule real-time (SMRT) sequencingProbes targeting CFH-CFHRs on the human genome, reference hg19 (from chr: 196619000 to chr: 196979303) were designed by Nimble Design Software (Roche Sequencing, Pleasanton, CA, US). Selected DNA samples previously identified with CNVs through MLPA analysis, were sequenced at the Norwegian Sequencing Centre using the PacBio Sequel system. Methodology details have previously been published in Piras R et al. (17)
Direct sequencingPCR was carried out in 25 µl of reaction volume using 125 ng of genomic DNA from patients with abnormal MLPA pattern and the Accuprime Taq DNA Polymerase (Thermo Fisher Scientific; 35 cycles of amplification: 94°C for 30 seconds, 59°C for 30 seconds, 68°C for 10 minutes). The breakpoint regions were identified by bidirectional sequencing of the long-range PCR product, using BigDye® Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific) following the manufacturer’s instructions. The BigDye XTerminator® Purification Kit (Thermo Fisher Scientific) was used to purify DNA sequencing reactions removing non-incorporated BigDye® terminators and salts. Sequencing analyses were carried out on the 48-capillary 3730 DNA Analyzer (Life Technologies). Sequences of primers used for long-range PCR are reported in Supplementary Table 1.
Western blotThe molecular pattern of FH-FHRs was studied by Western Blot (WB) using serum/plasma (diluted 1:40 for FHRs and 1:80 for FH). Proteins were separated by 10–12% SDS-PAGE, under non-reducing conditions and transferred by electroblotting to polyvinylidene Difluoride (PVDF) membrane (Bio-Rad). Membranes were developed using specific FH/FHR antibodies (the FHR-3 polyclonal antiserum was a kind gift from Prof. Zipfel (15); the anti-FHR1-2-5 monoclonal antibody was kindly provided by Prof. de Cordoba (12); the commercial monoclonal anti-human Factor H - OX-23, LSBio-), followed by HRP- conjugated secondary antibodies and ECL chemiluminescence detection system (Amersham).
Statistical analysisAll statistical tests were executed using MedCalc software. The Chi-square test or the Fisher’s exact test were used to make comparisons, as appropriate.
ResultsCFH-CFHR structural variants in aHUSWe report a retrospective MLPA analysis in a cohort of 350 unrelated patients with a diagnosis of aHUS, including 258 with primary aHUS and 92 with secondary aHUS.
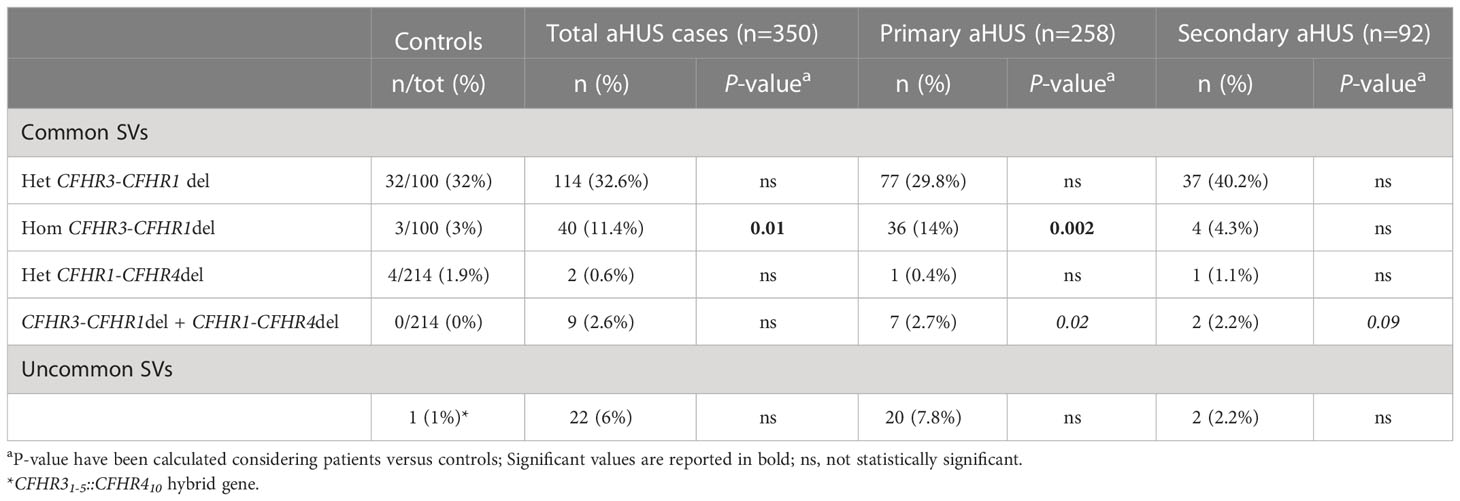
Common structural variants (SVs), namely the CFHR3-CFHR1 deletion (CFHR3-CFHR1del) and/or the CFHR1-CFHR4 deletion (CFHR1-CFHR4del), were observed in 165 patients (47%; Table 1). The homozygous CFHR3-CFHR1del was significantly more frequent in aHUS cases than in healthy controls (11% vs 3%, respectively, p-value = 0.01).
Table 1 Frequency of common and uncommon Structural Variants (SVs) in controls and patients.
The prevalence of common SVs was comparable in primary and secondary aHUS patients (47% vs 48%, respectively, ns), although the homozygous CFHR3-CFHR1del was more frequent in primary than in secondary aHUS (14% vs 4%, respectively, p-value = 0.01).
Specifically, in the primary aHUS group, common SVs were detected in 121 patients (47%) (Table 1): the heterozygous CFHR3-CFHR1del was found in 77 patients (30% vs 32% in healthy controls, ns); 36 patients exhibited the homozygous CFHR3-CFHR1del (14% vs 3% controls, p-value = 0.002); in addition, 7 patients were carriers of both CFHR3-CFHR1del and CFHR1-CFHR4del (3% vs 0% controls, ns) and a single case had the heterozygous CFHR1-CFHR4del (0.4% vs 2% controls, ns).
Among patients with secondary aHUS, common SVs were detected in 44 patients (48%). As shown in Table 1, 37 patients were heterozygous for the CFHR3-CFHR1del (40% vs 32% controls, ns) and 4 patients exhibited the same deletion on both alleles (4% vs 3% controls, ns). In addition, 2 patients were carriers of both the CFHR3-CFHR1del and the CFHR1-CFHR4del (2% vs 0% controls, p-value = 0.09) and one patient had the heterozygous CFHR1-CFHR4del (1% vs 2% controls, ns).
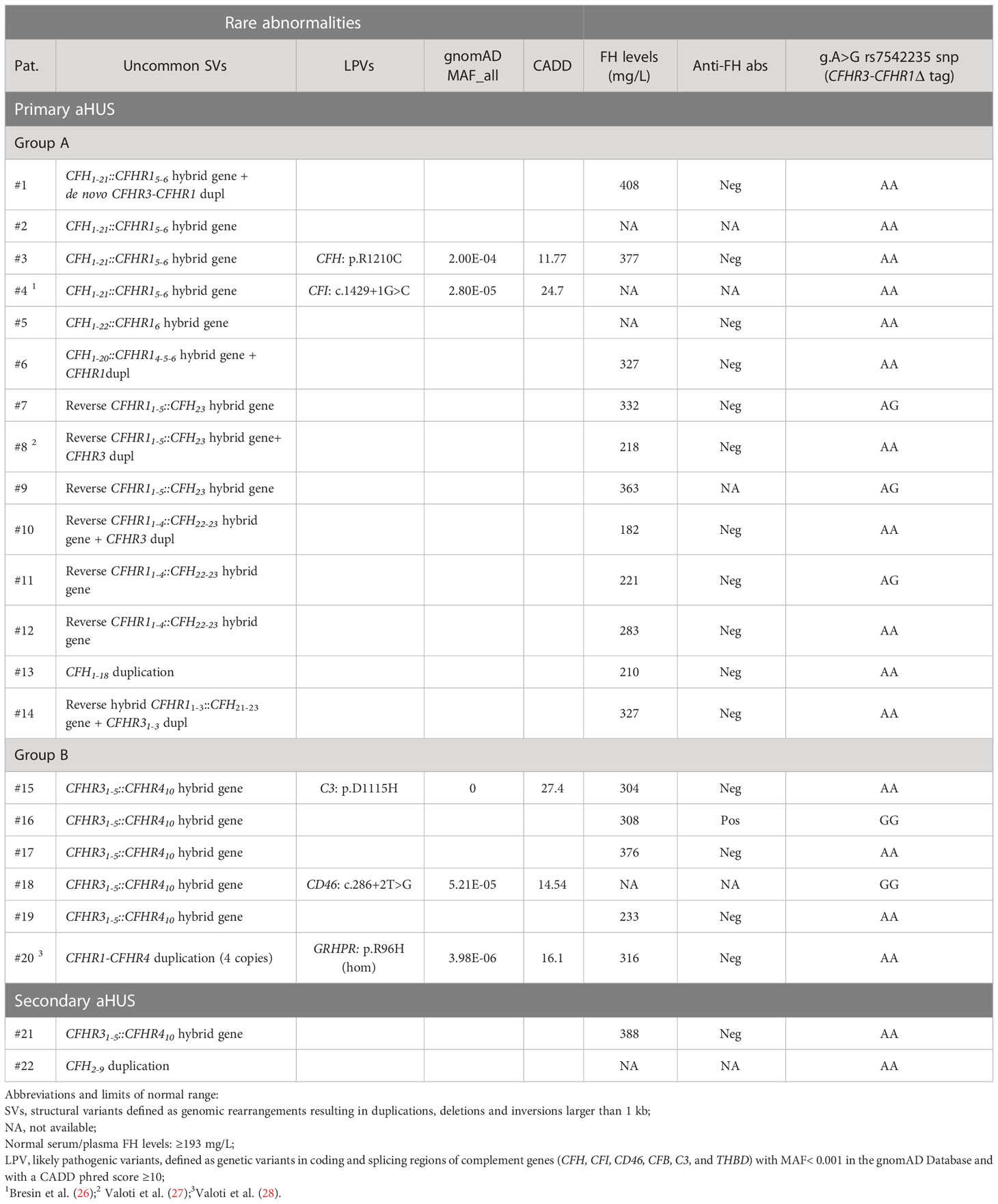
Twenty-two patients (6%) carried uncommon SVs, including new or rare duplications and hybrid genes. These uncommon SVs were mainly found in patients with primary aHUS (n=20 out of 22 carriers; 91%), with a prevalence in this group of 8%.
Seventy % are rearrangements involving CFH gene alone or CFH and CFHR genes (n=14) and 30% are rearrangements including only CFHR genes (n=6; Table 2) that were mainly reported in C3G (17).
Table 2 List of patients with new/rare CFH-CFHR structural variants (SVs) and/or other complement abnormalities.
From here on we divided patients with rearrangements in CFH alone or CFH and CFHR genes (group A) from those involving only CFHRs (group B) to investigate differences in prevalence, age of onset, outcome and response to therapy.
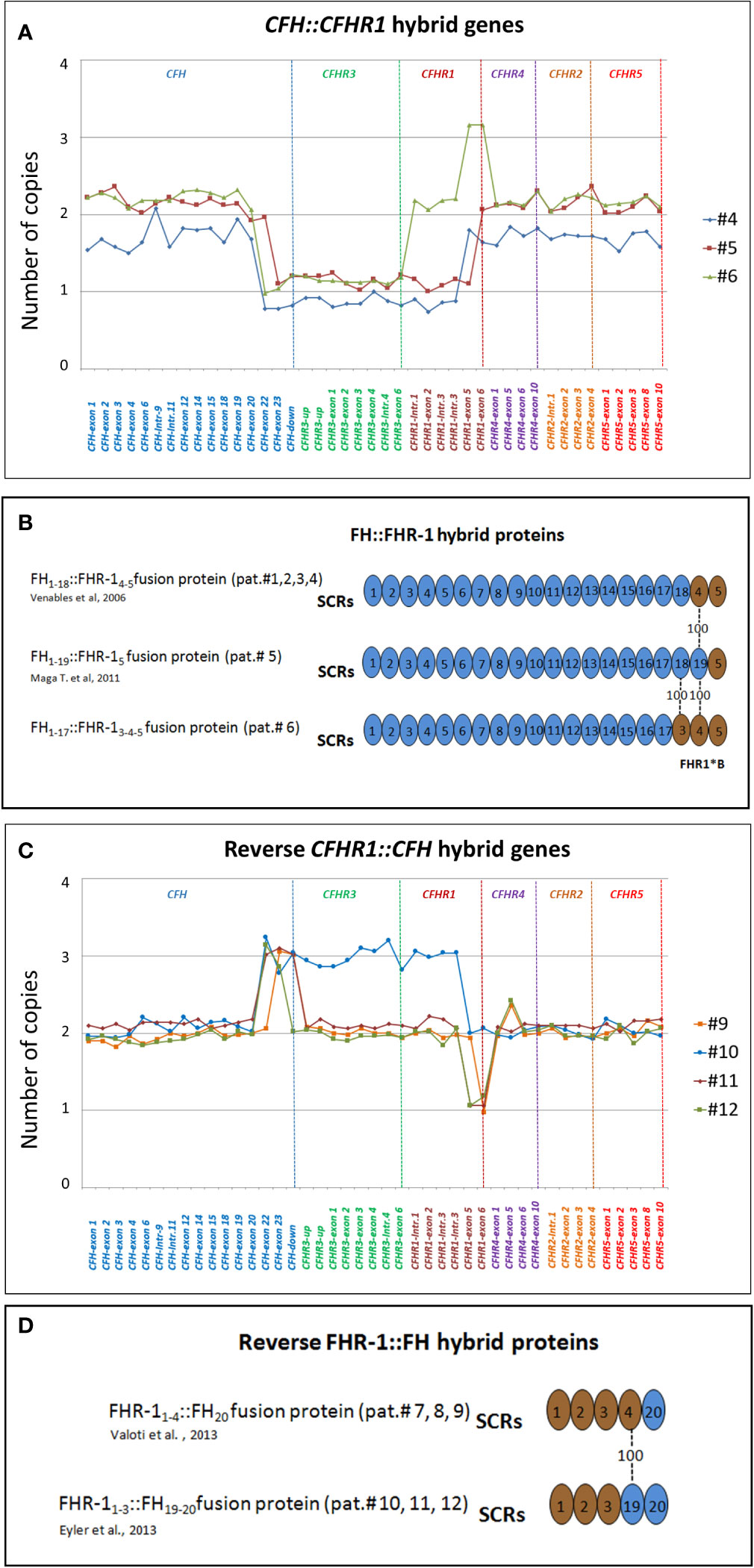
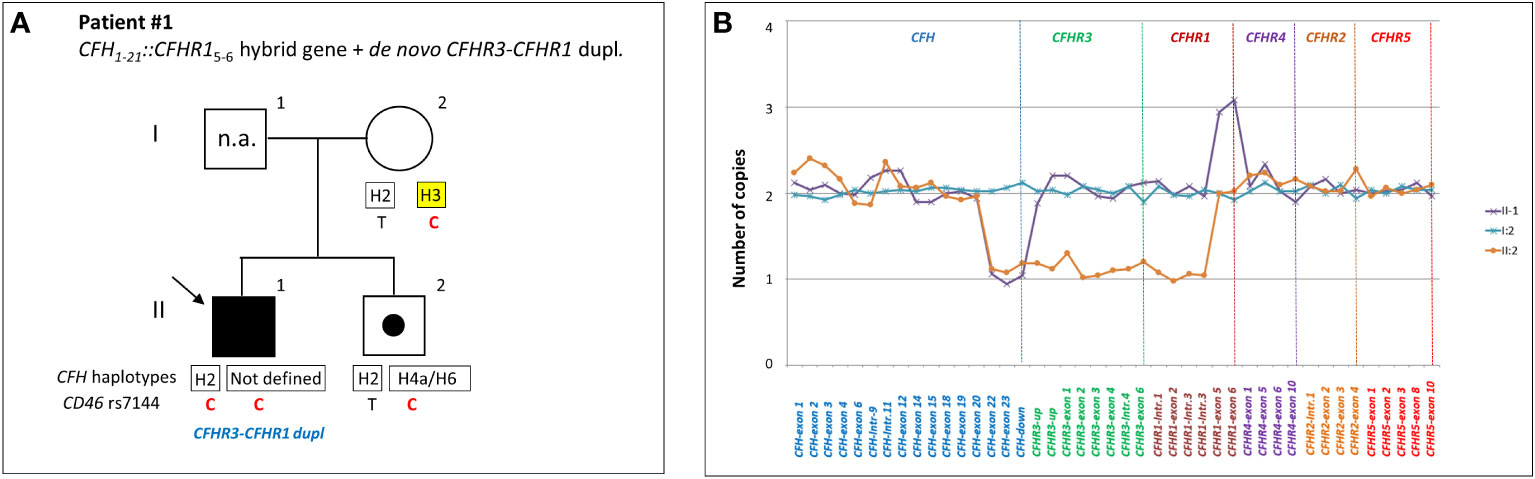
Primary aHUS group ACFH::CFHR1 hybrid genes: Six unrelated patients (#1; #2; #3; #4; #5; #6) shared a similar MLPA pattern in which probes showed one copy of exon 23 (n=1) or exons 22 and 23 (n=4) or exon 21-23 (n=1) of CFH and a gain of exon 6 or exons 5-6 or exons 4-6 of CFHR1, respectively (Table 2 and Figure 2A). In patients #1, #2, #3 and #4 the abnormal MLPA pattern is consistent with a deletion giving rise to CFH1-21::CFHR15-6 hybrid gene, described for the first time in a family in UK (29). Notably, patient #1 also exhibited an extra copy of both CFHR3 and CFHR1. CNV analysis of his relatives found the CFH1-21::CFHR15-6 hybrid gene in the patient and in his healthy brother – with the latter lacking the extra copy of CFHR3 and CFHR1– and a normal copy number in his healthy mother. Biological samples from the father were not available. Nonetheless, these results suggest that the patient inherited the hybrid CFH1-21::CFHR15-6 gene from his father and evidenced the presence of a de novo CFHR3-CFHR1 duplication (Figure 3). In patients #2, #3 and #4, the breakpoints of the CFH1-21::CFHR15-6 hybrid gene were located to different genomic positions within intron 21 (#2, #3 chr1: 196712875-196797547; #4 chr1: 196712997-196797845).
Figure 2 Graphic representation of MLPA results from aHUS patients carrying structural variants (SVs) in the CFH and CFHR genes. (A) MLPA patterns consistent with CFH::CFHR1 hybrid genes. In patient #4, MLPA pattern shows one copy of exons 22 and 23 of CFH, one copy of CFHR3, one copy of CFHR1 until intron 3, 2 copies of exons 5 and 6 of CFHR1 and two normal copies of CFHR4, CFHR2 and CFHR5, consistent with CFH1-21::CFHR15-6 hybrid gene (29). In patient #5 MLPA results evidence 1 copy of exon 23 of CFH, one copy of CFHR3, one copy of CFHR1 until exon 5, 2 copies of exons 6 of CFHR1 and two normal copies of CFHR4, CFHR2 and CFHR5, consistent with CFH1-22::CFHR16 hybrid gene (30). In patient #6 MLPA analysis shows one copy of exons 22 and 23 of CFH, one copy of CFHR3, normal copy number from the CFHR1-intron 1 to CFHR1-intron 3, 3 copies of exons 5 and 6 of CFHR1, and two normal copies of CFHR4, CFHR2 and CFHR5. This abnormal MLPA pattern was further characterized through long PCR, Sanger sequencing and SMRT which, as reported in Figure 3, led to the identification of the CFH1-20::CFHR14-6hybrid gene. (B) Representation of FH::FHR-1 hybrid proteins resulting from SVs identified in patients #1, #2, #3, #4, #5 and #6. SCRs translated from CFH are indicated in blue while SCRs deriving from CFHR1 are indicated in brown. The number “100” indicates that SCR4 of FHR-1 is identical to SCR19 of FH; similarly, SCR3 of FHR-1*B is identical to SCR18 of FH. The total identity between FH and FHR-1 indicates that the translated FH::FHR-1 protein is the same in all the above described cases. (C) MLPA pattern consistent with reverse CFHR1::CFH hybrid genes. MLPA results in patient #9 show 3 copies of CFH-exon 23, normal CFHR3 copies, two CFHR1 copies until exon 5, one copy of CFHR1-exon 6, two normal copies of CFHR4, CFHR2 and CFHR5, consistent with the reverse CFHR11-5::CFH23 hybrid gene. In patient #10, #11 and #12 MLPA analysis shows a gain starting from CFH-exon 22 until CFHR1-intron 3 and a loss of CFHR1-exon 5-6 consistent with a reverse CFHR11-4::CFH22-23 hybrid gene. In addition, patient #10 carries 3 copies of CFHR3 and 2 copies of normal CFHR1. In patient #11 MLPA provides a copy of normal CFHR1 and 2 copies of CFHR3, consistent with the presence of CFHR11-4-CFH22-23 on one allele and CFHR3-CFHR1 del on the other allele (see Supplementary Figure 1). Unlike patient #11, the abnormal MLPA pattern of patient #12 does not involve the probe located downstream of CFH. (D) Representation of reverse FHR-1::FH hybrid proteins resulting from SVs identified in patients #7, #8, #9, #10, #11 and #12. The translated fusion protein is the same in all cases due to the 100% of identity between SCR19-FH and SCR4-FHR-1.
Figure 3 Patient #1 with a hybrid CFH1-21::CFHR15-6 gene and a de novo CFHR3-CFHR1 duplication. (A) The proband (black arrow) is patient II:1, his father is I:1 (samples are not available, n.a.), his mother is I:2 and his brother (unaffected carrier of hybrid CFH1-21::CFHR15-6 gene) is II:2. Genotype of CFH single nucleotide polymorphisms (snps) targeting the CFH-H3 risk (TGTGT) haplotype (c.1–331C>T, rs3753394; c.184G>A, p.V62I, rs800292; c.1204T>C, p.Y402H, rs1061170; c.2016A>G, p.Q672Q, rs3753396; c.2808G>T, p.E936D, rs1065489) and the CD46 snp (rs7144, c.*897 T>C) targeting the CD46GGAAC risk haplotype are reported with a yellow square and in red, respectively. (B) MLPA analysis over the CFH-CFHR region in proband’s relatives shows three different patterns. Patient (II:1) exhibits one copy of CFH exons 22 and 23 and 3 copies of CFHR1 exons 5 and 6. His brother (II:2) exhibits a large heterozygous deletion from CFH exon 22 to CFHR1 intron 3, consistent with the presence of hybrid CFH1-21-CFHR15-6 gene. The mother (I:2) has a normal copy number. These results suggest that the patient and his brother inherited the hybrid CFH1-21::CFHR15-6 gene from their father and evidenced the presence of a de novo CFHR3-CFHR1 duplication only in the patient.
Patient #4 is a familial aHUS case with an affected first cousin (III-4; Supplementary Figure 1). MLPA studies revealed the CFH1-21::CFHR15-6 hybrid gene in both patients and in three unaffected family members (26).
In patient #5 we identified the same CFH1-22::CFHR16 genomic rearrangement described by Maga et al., which encodes the same fusion protein as the CFH1-21::CFHR15-6 hybrid gene (FH1-19::FHR-15 and FH1-18::FHR-14-5 respectively, Figure 2B) (30).
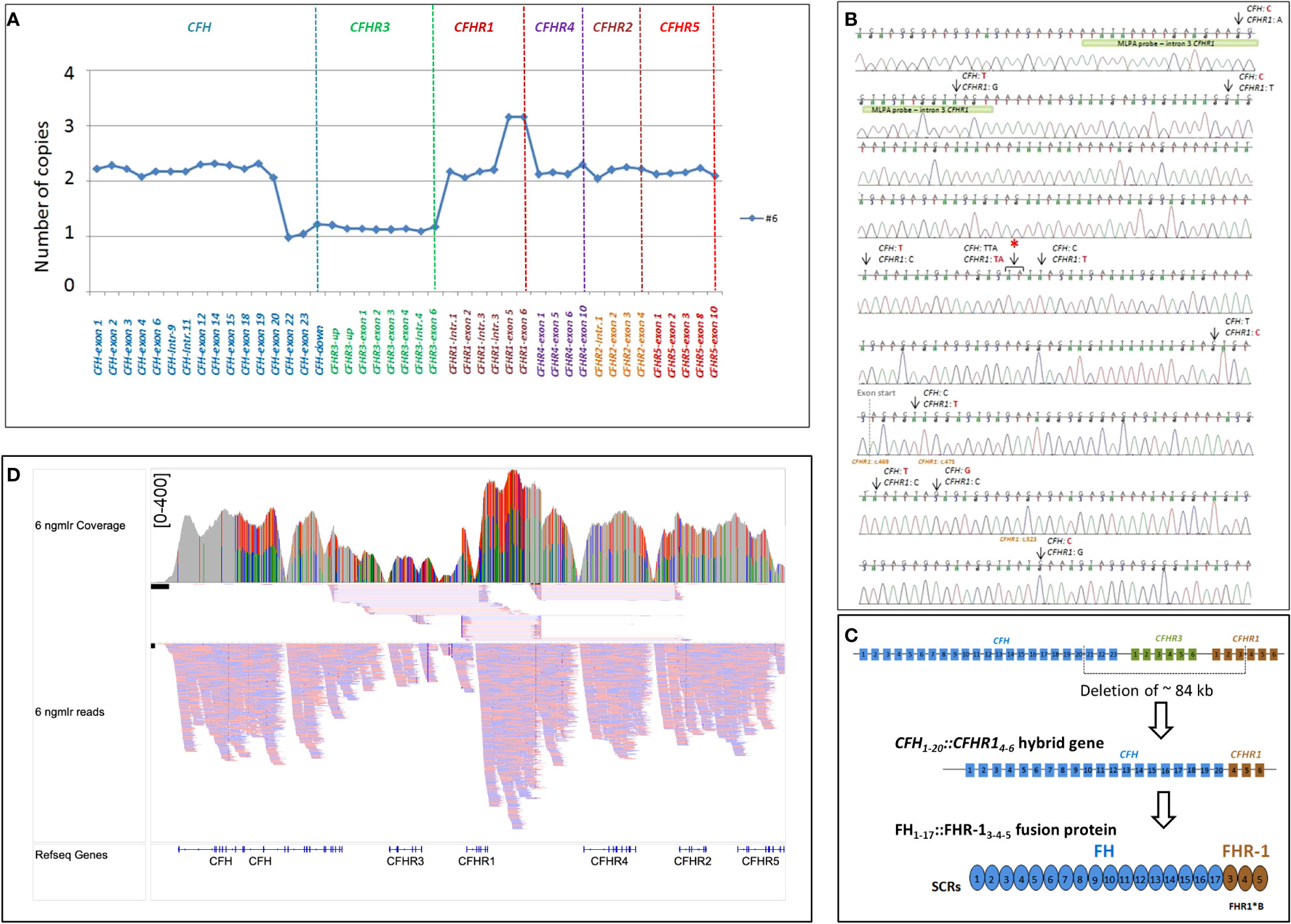
As shown in Figure 4, in patient #6, a large deletion extending from intron 20 of CFH to intron 3 of CFHR1 was identified by MLPA and long PCR followed by Sanger sequencing (Figures 4A, B), indicating a novel CFH1-20::CFHR14-6 hybrid gene, which resulted in a FH1-17::FHR-13-5 protein (Figure 4C). Since SCR3 and SCR4 of FHR-1 are identical to SCR18 and SCR19 of FH, the FH1-17::FHR-13-5 protein is indistinguishable from FH1-19::FHR-15 and FH1-18::FHR-14-5 (Figure 2B). The genomic breakpoints were mapped between chr1: 196712504 (intron 20 of CFH) and chr1: 196797138 (intron 3 of CFHR1). The identification of the CFH1-20::CFHR14-6 hybrid gene, confirmed also by SMRT sequencing (Figure 4D), did not fully clarify the abnormal CFHR1 MLPA pattern, characterized by the presence of 2 copies of intron 1, exon 2 and intron 3 and 3 copies of exons 5-6 of CFHR1. Together, these data indicated the presence of an extra copy of CFHR1.
Figure 4 Identification of CFH1-20::CFHR14-6 hybrid gene in patient #6. (A) The abnormal MLPA pattern found in patient #6 involves CFH, CFHR3 and CFHR1 genes. It results in: 1) loss of one copy of exons 22-23 of CFH; 2) loss of 1 copy of the entire CFHR3 gene; 3) 2 copies of intron 1-2 and 3 of CFHR1; 4) 3 copies of exon 5 and 6 of CFHR1. (B) Electropherogram including the sequence of the genomic breakpoint. Arrows indicate the nucleotide differences between CFH and CFHR1. The first part of the sequence corresponded to CFH; the red asterisk indicates the genomic position where the intron 4 CFHR1 sequence started. Four nucleotide differences were found in exon 4 CFHR1: c.469, c.475, c.523 (indicated in brown) and c.588 (not reported). Three of them led to three FHR-1 amino acid changes (p.Tyr157-Val159-Gln175, respectively) and are characteristic of the basic isoform of CFHR1 (CFHR1*B). The green bar highlights the target sequence of the “CFHR1-intron 3” probe, located in intron 3 of CFHR1 (194 nucleotides before exon 4), upstream of the breakpoint region, explaining the 2 copies identified by this probe. (C) Representation of ~84 kb deletion involving CFH, CFHR3 and CFHR1, resulting in the generation of CFH1-20::CFHR14-6 hybrid gene that encodes the FH1-17::FHR13-5 fusion protein. (D) Screenshot from IGV (Integrative Genomics Viewer) showing reads from SMRT sequencing. SMRT sequencing identified both CFH1-20::CFHR14-6 hybrid gene and the CFHR1 duplication. Misaligned reads in the CFHR1-CFHR4 intragenic region and the CFHR2 are also shown.
Results of WB analysis of patients’ plasma/serum are shown in Supplementary Figure 2A.
Reverse CFHR1::CFH genes: In patients #7, #8 and #9, the exon 6 of CFHR1 was replaced by the exon 23 of CFH, generating a reverse CFHR11-5::CFH23 hybrid gene, in addition to the normal CFHR1 (Figure 2C). As we showed in a previously published study, we also observed an extra copy of CFHR3 in patient #8 (27). Both the CFHR11-5::CFH23hybrid and the extra CFHR3 genes were transmitted to his progeny: his daughter developed aHUS, while his son was an unaffected carrier (Supplementary Figure 1). We previously showed that both these abnormalities were the result of a genomic duplication (27).
Patients #7 and #9 showed 3 copies of CFH-exon 23 and CFH-downstream probes and one copy of the CFHR1-exon 6 probe, consistent with the reverse CFHR11-5::CFH23 hybrid gene, but they had a normal CFHR3 and CFHR1 copy number. The presence in both patients of the heterozygous rs7542235 snp that tags the CFHR1–CFHR3 deletion suggests the presence of the reverse CFHR11-5::CFH23hybrid gene, with a normal CFHR1 copy and the extra CFHR3 copy on one allele and the common CFHR3-CFHR1del on the other allele (31).
In patients #10, #11 and #12, exons 5 and 6 of CFHR1 were replaced by exons 22 and 23 of CFH (Reverse CFHR11-4::CFH22-23 hybrid gene; Figure 2C). Similar to patient #8, in patient #10 we also identified a third copy of CFHR3 and 2 copies of CFHR1, as a result of a large genomic duplication.
The MLPA pattern of patient #11 was consistent with the extra reverse CFHR11-4::CFH22-23 hybrid gene, two copies of CFHR3 and one copy of CFHR1 (Figure 2C). The pedigree study showed that the proband inherited the reverse CFHR11-4::CFH22-23 hybrid gene from his mother and the common CFHR3-CFHR1del from his father (Supplementary Figure 1), indicating that the allele with the reverse hybrid carries a large genomic duplication, involving CFHR3, as reported in patient #10.
A similar but not identical MLPA pattern (not involving the probe located downstream of CFH; Figure 2C) was observed in patient #12. In both patients #11 and #12, Sanger sequencing placed the genomic breakpoints between intron 4 of CFHR1 and intron 21 of CFH, confirming a reverse CFHR1-CFH gene, but the genomic locations were different (#11: chr1: 196799104-196714588; #12: chr1: 196797930-196713442). Patient #12 did not carry the rs7542235 snp linked to the CFHR3-CFHR1 deletion, which does not indicate the presence of a deletion on the other allele, unlike patient #11. Altogether these data show that the reverse hybrid in patients #11 and #12 could derive from different genomic rearrangements. A schematic representation of the reverse FHR-1::FH proteins is shown in Figure 2D. Results of WB analysis of patients’ plasma/serum are shown in Supplementary Figure 2B.
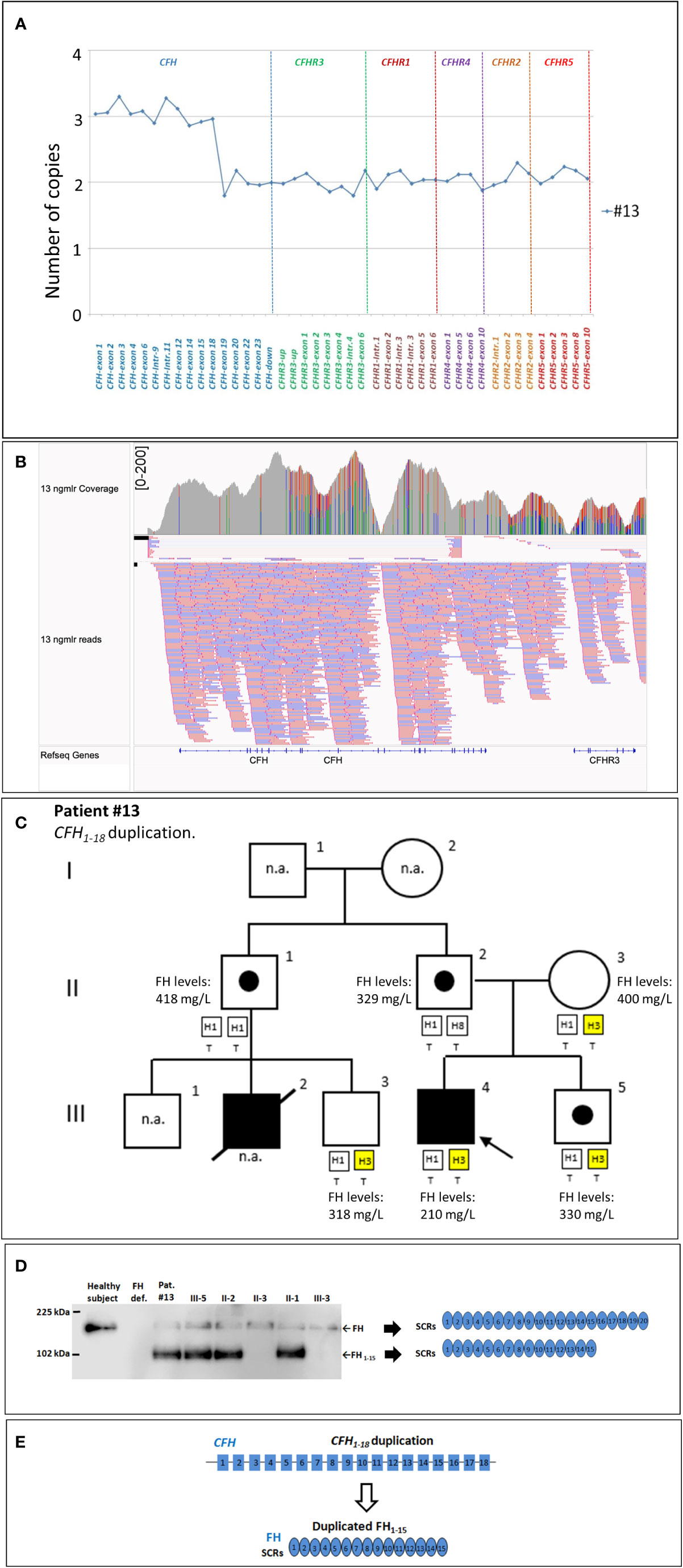
New genomic rearrangements: We identified a 98 kb tandem duplication in CFH extending from exon 1 to exon 18 (chr1: 196611131-196708834) in a single case affected by a primary form of aHUS (#13; Figures 5A, B). The duplication was inherited from the unaffected father (II-2) and was also found in his healthy brother (III-5) and in an unaffected paternal uncle (II-1; Figure 5C). Their family history shows that a paternal cousin (III-2; son of II-1) had recurrent aHUS and died at 10 years of age. FH levels in the proband and in the available relatives were normal (≥193 mg/L) although the proband had lower FH levels (210 mg/L) than his relatives (Figure 5C). WB using a monoclonal anti-human FH antibody and samples from all carriers of the CFH1-18 duplication showed: 1) the band of normal FH protein, around 155 kDa; 2) a shorter than normal band with a MW around 100 kDa (Figure 5D). These results indicate that the CFH1-18 duplication produces a short FH, likely composed by the first fifteen SCRs of FH (FH1-15) (Figure 5E).
Figure 5 The CFH1-18 duplication identified in patient #13. (A) MLPA pattern showing 3 copies of CFH until exon 18 and 2 normal copies in the remaining CFH, CFHR exons. (B) Screenshot from IGV (Integrative Genomics Viewer) showing reads from SMRT sequencing of patient #13, carrying a tandem CFH1-18 duplication. Reads originating from across the breakpoint were mapped as chimeric alignments (split-reads) with the second part of the read mapped upstream of the first part (and vice versa for the reverse reads). (C) Pedigree of patient #13 and FH levels: the CFH1-18 was inherited from the unaffected father and was also found in both his healthy brother (III-5) and in unaffected paternal uncle (II-1). FH levels resulted in the normal range in all tested samples (n.r.: ≥193 mg/L) although in the proband’s sample were lower (210 mg/L) than in the other relatives. (D) Western Blot (WB) to detect FH was performed using a monoclonal anti-human FH antibody (OX-23, LSBio), under non-reducing conditions, using sample from the proband (III-4), his available relatives, a patient with FH deficiency (negative control) and a healthy control with normal FH (positive control). The presence of a band with a MW (around 100 kDa) lower than normal FH in all carriers of the CFH1-18 duplication indicates that a short FH, likely missing the C-terminal domains [FH1-15; (E)], is secreted. n.a., not available.
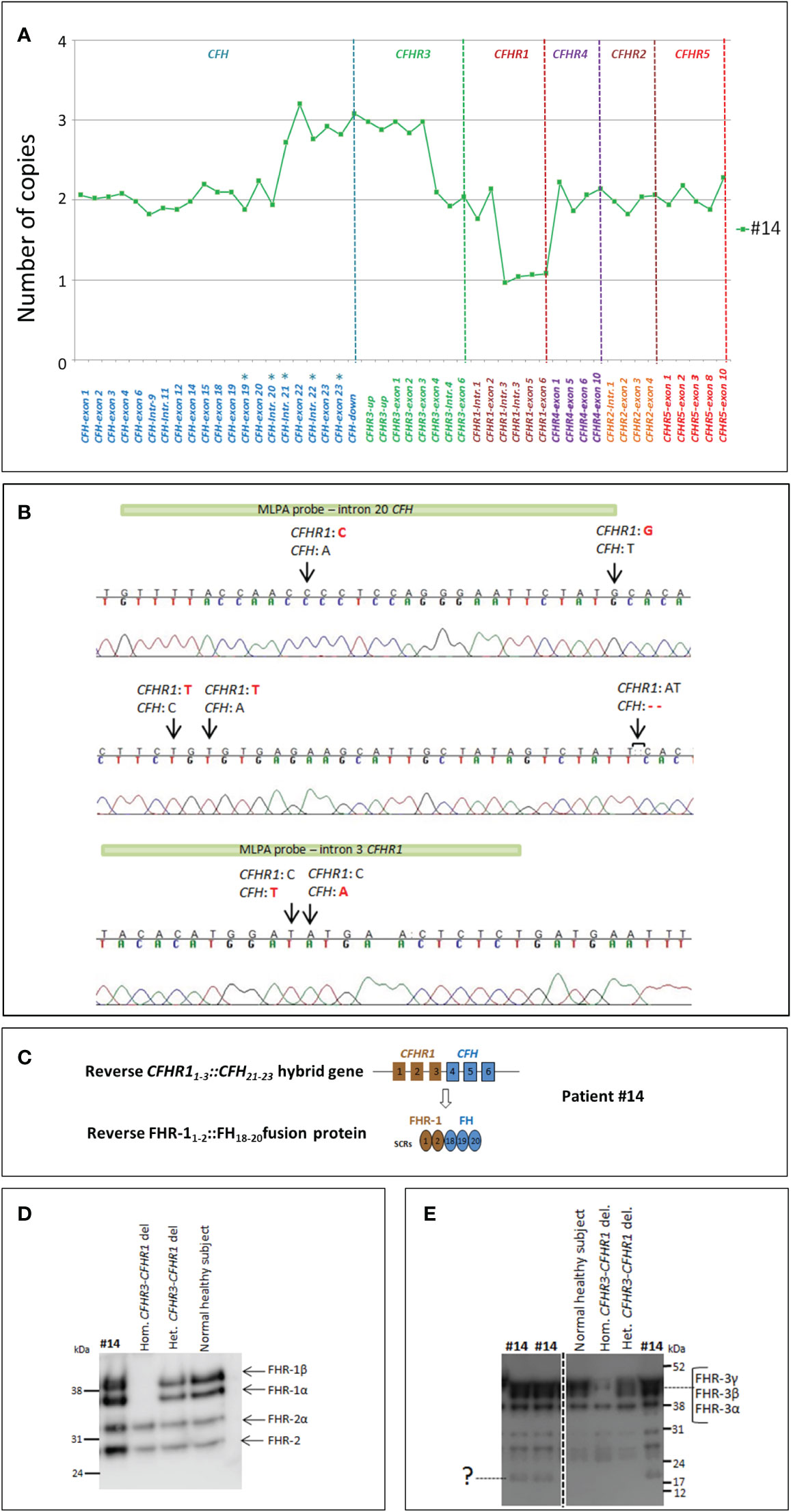
An additional new SV was observed in patient #14, characterized by 3 copies of CFH intron 21-exons 22-23 and CFHR3 exons 1-2-3, with the concomitant presence of 2 copies of CFHR1, one of them lacking exons 4-5-6 (Figure 6A). PCR, using a forward primer located in intron 2 of CFHR1 and a reverse primer located in intron 21 of CFH, and Sanger sequencing revealed the presence of a CFHR11-3::CFH21-23 hybrid gene, likely resulting in a reverse FHR-11-2::FH18-20 (Figures 6B, C). Western Blot analysis using an anti-FHR-1-2-5 antibody did not discriminate between the wt FHR1 and the reverse hybrid FHR-11-2::FH18-20 (same molecular weight, Figure 6D). Analysis using an anti-FHR-3 antiserum revealed in the serum from patient #14: 1) three bands corresponding to normal glycosylated isoforms of FHR-3 (Figure 6E and Supplementary Figure 2C); 2) a single band at lower MW (around 15 kDa; Figure 6E) compatible with a shorter FHR-31-2 protein.
Figure 6 Identification of the reverse CFHR11-3::CFH21-23 hybrid gene in patient #14. (A) The results of MLPA show in patient #14 three copies of both CFH exons 21-22-23 and CFHR3 exons 1-2-3, 2 copies of CFHR1, one of them lacking exons 4-5-6. These data suggest the presence of a reverse CFHR11-3::CFH21-23 hybrid with a partial duplication of CFHR31-3. The analysis has been performed with the SALSA MLPA P236-CFH-region Kit (MRC Holland) implemented with homemade probes (indicated by asterisks) analyzed in a separate assay and covering the last exons and introns of the CFH gene (27). (B) Sequence of the genomic breakpoint of the CFHR11-3::CFH21-23 hybrid gene, mapped between chr1:196796490 (intron 3 of CFHR1) and chr1:196711901 (intron 20 of CFH). Arrows indicate the nucleotide differences between CFH and CFHR1. The green bars highlight the target sequence of “CFH-intron 20” probe (located 747 nucleotides before exon 21) and the “CFHR1-intron 3” probe (located 396 nucleotides after exon 3). (C) Representation of reverse CFHR11-3::CFH21-23 and the corresponding FHR-11-2::FH18-19-20 fusion protein. (D, E) To investigate the effect of CFHR31-3 duplication at protein level, we performed a WB analysis, under non-reducing conditions, using both an anti-FHR-1-2-5 monoclonal antibody and a FHR-3 polyclonal antiserum. FHR-1 staining showed FHR-1 bands at the same MW of the normal healthy control (D). Staining of FHR-3 showed both the bands corresponding to the three normal glycosylated isoforms of FHR-3 and a faint band at low MW consistent with a short FHR-31-2(E).
Primary aHUS group BCFHR31-5::CFHR410 hybrid gene and CFHR1- CFHR4 duplication: This group was characterized by uncommon genomic rearrangements involving only CFHR genes (Table 2).
In five patients (#15, #16, #17, #18, and #19) we identified the CFHR31-5::CFHR410 hybrid gene that we previously described in C3G (17). Among these, case #16 also carried the polymorphic CFHR3-CFHR1del, leading to the lack of CFHR1.
In this group the only available pedigree was that of patient #15. MLPA studies revealed the presence of the CFHR31-5::CFHR410 hybrid gene in his healthy mother, too (Supplementary Figure 1). Results of WB analysis of patients’ plasma/serum are shown in Supplementary Figure 2D.
Finally, four copies of both the CFHR1 and CFHR4 genes were observed in patient #20, a previously described case of aHUS concomitant to primary hyperoxaluria due to GRPHR gene abnormalities (Supplementary Figure 1) (28).
Genetic and serum abnormalities in primary aHUSTo better characterize patients with uncommon SVs, we also evaluated the presence of LPVs in complement genes and or/anti-FH antibodies (anti-FHs).
We observed that group B had a higher prevalence of concomitant complement abnormalities (4/6) compared to group A (2/14) even though the difference was not statistically significant (p-value= 0.04).
In detail, only two patients out of 14 (14%; patients #3 and #4) of group A, both carrying the CFH1-21::CFHR15-6 hybrid gene, had additional complement abnormalities (CFH p. R1210C and CFI c.1429+1G>C LPVs, respectively).
In group B, three of six patients (50%; patients #15, #16 and #18), all carrying the CFHR31-5::CFHR410 hybrid gene, also had concomitant LPVs (#15: C3 p.D1115H and #18: CD46 c.286+2T>G, respectively) or anti-FHs (patient #16, with the CFHR31-5::CFHR410 hybrid gene on one allele and the CFHR3-CFHR1del on the other allele) (Table 2).
To evaluate whether the presence of SVs impaired circulating factor H (FH) levels we tested serum/plasma FH concentrations in all patients carrying uncommon SVs, for whom samples were available. Only patient #10, with the reverse CFHR11-4::CFH22-23 hybrid gene (group A), had FH levels that were slightly lower than normal (182 mg/L; n.v.≥ 193 mg/L), indicating that CFH-CFHR SVs did not substantially impact FH levels (Table 2).
Finally, to evaluate whether in our cohort of primary aHUS patients, the complete CFHR1 deficiency was associated with the presence of anti-FHs, patients carrying the homozygous CFHR3-CFHR1del or the combined CFHR3-CFHR1del and CFHR1-CFHR4del were tested for anti-FHs, when samples were available. We found that 26 out of 39 tested patients had anti-FHs (67%), consistently with the already known correlation between CFHR1 deficiency and development of anti-FHs in patients with aHUS (3, 18).
Incomplete penetrance of rare structural variantsAmong the nine studied pedigrees, we found 17 asymptomatic relatives carrying rearrangements in CFH or CFHR genes, indicating that SVs are associated with an incomplete penetrance of the phenotype aHUS (11 affected/28 carriers; 39%).
As reported above, in patients #3, #4, #15, #16 and #18 we identified additional complement abnormalities. Samples from relatives were available for patients #4 and #15 only. In the pedigree of patient #4, the CFI LPV (c.1429+1G>C) was found in the proband (IV-3), but also in her healthy mother (III-1) and in the younger sister (IV-4), who do not carry the CFH1-21::CFHR15-6 hybrid gene (Supplementary Figure 1). These results suggested that the concomitant presence of SVs and LPVs synergized in determining disease development in the proband. Accordingly, her father (III-2) and the older sister (IV-2) carried only the CFH1-21::CFHR15-6 hybrid gene and did not have aHUS. However, the CFH1-21::CFHR15-6 hybrid gene but not the CFI LPV (c.1429+1G>C), were also found in a healthy grand-uncle of the proband (II-3) and his daughter (III-4), who developed ESRD after an episode of aHUS. These findings indicate that in this arm of the pedigree, other risk factors synergized with the CFH1-21::CFHR15-6 hybrid to the final phenotype. As shown in Supplementary Figure 1, the affected subject in this arm (III-4) is homozygous for the CD46GGAAC risk haplotypes, whereas her unaffected father is heterozygous.
Incomplete penetrance was also observed in the pedigree of patient #15 carrying the CFHR31-5::CFHR410 hybrid gene, the C3 LPV (p.D1115H), and the CFH-H3 risk haplotype, all inherited from the unaffected mother (Supplemantary Figure 1).
Clinical data of patients with primary aHUS carrying rare SVsIn group A, infectious triggers were reported in all patients with available data (7/7) and the median age at disease onset was 6.5 years (IQR, 1-25.75). Two out of 14 patients were treated with eculizumab: one of them (patient #9) underwent full remission with complete recovery of renal function, the other (patient #7) had an aHUS relapse in the kidney graft. Eculizumab treatment enabled stable normalization of hematological parameters and partial recovery of graft function, but thereafter the patient lost the graft due to chronic rejection. In contrast, 11 out of 12 patients in this group who did not receive eculizumab (either because disease onset antedated anti-complement therapy or the drug was not available) did not recover from the acute episode and developed end-stage renal disease (ESRD; Table 3).
Table 3 Demographic and clinical findings of aHUS patients carrying uncommon CFH-CFHR SVs.
Atypical HUS relapses occurred in 6/7 grafts without eculizumab prophylaxis and in 0/3 grafts with eculizumab prophylaxis.
In group B, infectious triggers were associated with aHUS onset in 5/5 cases with available data, and the median age of disease onset was 2 years (IQR, 1.5-7.5).
Disease was less severe in group B than in group A. Indeed, 4 out of 5 patients in this group achieved complete remission without eculizumab treatment (p=0.0099, vs group A no eculizumab). No clinical data are available for patient #19.
One group B patient (patient #15) lost two kidney grafts for aHUS recurrence; he did not receive eculizumab (Table 3).
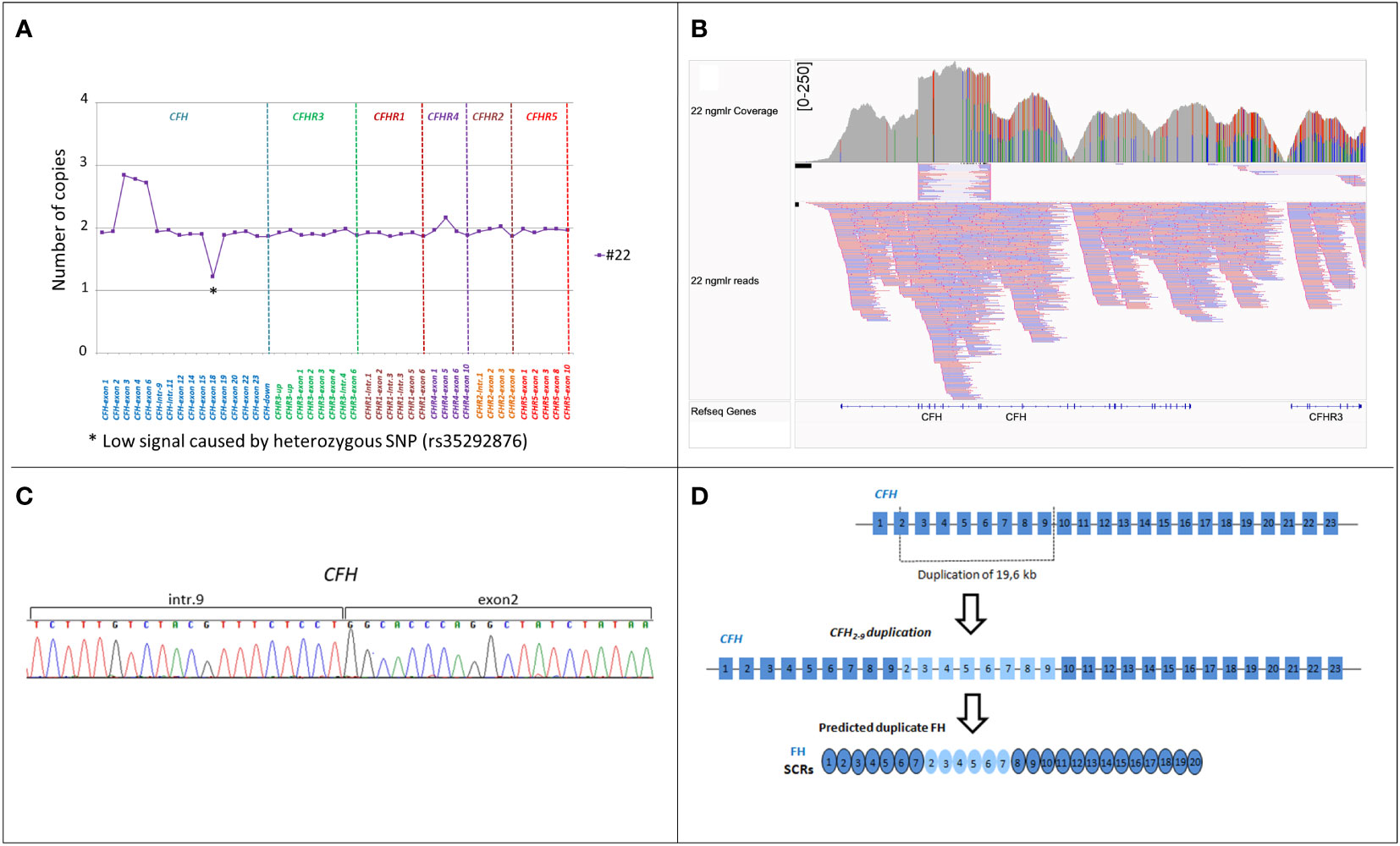
Rare structural variants and clinical data of patients with secondary aHUSTwo cases of secondary aHUS, associated with malignant and severe hypertension, respectively, had uncommon SV (#21; #22; Table 2). One of them carried the hybrid CFHR31-5::CFHR410 gene, previously described in a patient with DDD (17) while the other patient had an internal duplication in CFH, extending from part of exon 2 to exon 9 (Figure 7A). Direct sequencing of a long-PCR product and SMRT sequencing allowed us to map the duplication within CFH (chr1:196642182-196661791; Figures 7B, C) with partial intron 9 sequence followed by part of exon 2 sequence. In silico analysis (Genscan) predicted that the CFH2-9 duplication may generate a longer CFH gene, characterized by the first nine exons of CFH, followed by exon 3 of CFH (Figure 7D).
Figure 7 Identification of CFH2-9 duplication in patient #22. (A) MLPA pattern in patient #22 shows a high signal on the exon 3, exon 4 and exon 6-CFH probes consistent with a heterozygous duplication. (B) Screenshot from IGV (Integrative Genomics Viewer) showing reads from SMRT sequencing. SMRT sequencing identified CFH2-9 duplication. (C) Electropherogram of the genomic breakpoint. The first part of the sequence corresponds to intron 9 of the CFH and the second part is the sequence of CFH exon 2. (D) Representation of 19,6 kb internal duplication of the CFH and the predicted resulting FH protein consisting of 26 SCRs.
In both patients no LPVs in complement genes were identified (Table 2).
The outcome of the patient with the hybrid CFHR31-5::CFHR410 gene (#21) was unfavorable as she developed ESRD (no eculizumab treatment; Table 3). The patient carrying the internal duplication of CFH was treated with eculizumab, which partially improved renal function and hematological parameters at one month after disease onset (Table 3).
DiscussionIn the present study, through a retrospective analysis of a large cohort of patients affected by either primary or secondary forms of aHUS, we documented that: I) the prevalence of the homozygous CFHR3-CFHR1del is higher in primary aHUS than in secondary aHUS; II) uncommon SVs in CFH and CFHR genes are more frequent in primary aHUS than in secondary forms; III) the disease penetrance of aHUS in carriers of rare CFH-CFHR SVs is incomplete; IV) these genomic abnormalities occur often in combination with other complement abnormalities; V) the prognosis for rare SV carriers is strongly related to the specific abnormality.
Among common SVs, we observed an enrichment of the homozygous CFHR3-CFHR1del in primary aHUS and confirmed previous data in the literature about the association between this common SV and the development of anti-FH autoantibodies, an acquired driver reported in 10% of aHUS patients (3, 18). In contrast, the finding that in secondary aHUS patients the prevalence of the homozygous CFHR3-CFHR1del was comparable to controls does not support the hypothesis that this deletion plays a role in secondary aHUS. Similarly, we identified rare SVs, including duplications or hybrid genes, in a substantial fraction (8%) of patients with aHUS, but we rarely did so in secondary forms (2%).
Consistent with earlier data in literature, in our cohort uncommon SVs most frequently involved CFH and CFHR1 leading to the formation of CFH::CFHR1 hybrid genes or reverse CFHR1::CFH hybrid genes (16, 27, 29, 30, 32).
Specifically, we identified the CFH1-21::CFHR15-6 and the CFH1-22::CFHR16SVs in 2% of patients with primary aHUS. Even though they formed through different rearrangements, as documented by the identification of different DNA breakpoints, hybrid CFH::CFHR1 genes caused the loss of one copy of CFHR3 and encoded for the same FH::FHR-1 fusion protein characterized by a FH protein in which SCR19 and 20 were substituted by the FHR-1 specific SCR4-SCR5 C-terminal residues (29, 30). Notably, the FH SCR19 is identical to FHR-1 SCR4, while FH SCR20 differs from FHR-1 SCR5 only at 2 amino acids: FH Ser1191 (which corresponds to Leu290 in FHR-1) and FH Val1197 (corresponding to Ala296 in FHR-1). Thus, the FH-FHR-1 fusion proteins encoded by the CFH1-21::CFHR15-6 and the CFH1-22::CFHR16 hybrids share the FHR-1-specific C-terminus with the Leu1191 and Ala1197 changes. As reported in published studies, the FH with 1191Leu and 1197Ala residues can also derive from a gene conversion event that is the result of the unidirectional transfer of CFHR1 exon 6 into the CFH gene (33, 34). Functional studies have shown that these FH mutants have a normal regulatory activity in the fluid phase but have a limited capacity to protect cells from complement activation (33). This is also consistent with recent findings showing that the two amino acid differences that differentiate the C-terminus of FH from that of FHR-1 are responsible for distinctive interactions with C3 and sialic acid glycans (35, 36). Indeed, while the C-terminus of FH binds to proteoglycans and C3b, thus driving the recruitment of FH to cell surfaces and to the cell matrix, the C-terminus of FHR-1 strongly interacts with native C3 (nC3), C3b, iC3b and C3dg, attracting nC3 to the proximity of the cell surface. The latter acquired property of FH::FHR-1 fusion proteins, along with the loss of the capacity to bind sialic acids, causes a shift from complement regulation to complement activation on cell surfaces (35).
Another CFH and CFHR1 genomic rearrangement reported in association with aHUS leads to reverse CFHR1::CFH genes. A de novo CFHR11-4::CFH22-23 gene was described in 2013 in a patient with sporadic aHUS, and in 2015 we reported a reverse CFHR11-5::CFH23 gene in a family with 2 affected subjects over 2 generations (27, 32). Here, we identified 5 additional patients carrying the above reverse CFHR1::CFH genes, which derived from different DNA breakpoints. Notably, all these genes encode the same FHR-1::FH fusion protein characterized by the FH specific C-terminus with amino acid 290Ser (which corresponds to Ser1191 in FH) and 296Val (that corresponds to Val1197 in FH) (37). The reverse FHR-1::FH hybrid, through its FHR-1 N-terminal domain forms multimers that interact with other FHR-1 and/or FHR-2 molecules, while with its FH C-terminal domain competes with FH for the binding to cell ligands, thus promoting surface restricted complement activation (16, 35). Consistently, FHR-1 isolated from heterozygous carriers of the CFHR11-5::CFH23 hybrid induced complement-dependent sheep erythrocytes hemolysis when added to normal human serum (27).
In this study, we have also identified a new reverse CFHR11-3::CFH21-23 gene including a partial duplication of the first three exons of CFHR3 likely resulting in a FHR-11-2-FH18-20 protein and in a short FHR31-2 protein that requires further investigation. We speculate that the greater sequence similarity to FH, makes the FHR-11-2-FH18-20 protein a stronger competitor of FH for its surface ligands, than the reverse FHR-11-4::FH20 and FHR-11-3::FH19-20 hybrids.
Another novel finding of this study is the association of large CFH gene duplications with aHUS. In a 10-year boy with primary aHUS, we identified a CFH tandem duplication involving a large portion of the gene, including exons 1 to 18, and encoding a shorter than normal FH (likely FH1-15) that lacks the five C-terminal SCRs. We speculate that the abnormal FH cannot properly bind and inhibit AP on cell surfaces while maintaining its inhibitory functions in fluid phase. In addition, in a 22-year-old woman with secondary aHUS associated with severe hypertension, we found an internal CFH duplication extending from part of exon 2 to exon 9 and located after CFH intron 9. We hypothesize that similarly to partial duplications described in other genes, this CFH duplication may either cause a reading frame shift in the mRNA, producing a truncated FH, or result in a longer than normal protein with conformational changes and dysfunctional activity (38–40). Consistently, in silico analysis of the CFH2-9 duplication predicts a longer translated FH product (Figure 7D).
Notably, we observed incomplete penetrance of aHUS in carriers of CFH-CFHRs SVs, which is consistent with earlier studies on patients with LPVs in CFH and other complement genes (23, 41, 42). The identification of additional rare complement gene abnormalities in 3 probands with CFH-CFHRs SVs is in line with the above observation. Specifically, in a pedigree with the CFH1-21::CFHR15-6 hybrid gene we found a CFI variant, predicted to alter splicing in the region encoding serine protease domain of Factor I, in the proband who developed aHUS in the first year of life, whereas this variant was absent in all unaffected carriers of the hybrid gene. However, the finding that in this family a carrier of the CFH1-21::CFHR15-6alone developed aHUS, whereas there were none among the subjects with the CFI LPV alone, documents that the CFH1-21::CFHR15-6hybrid gene is the main driver of the disease. In a patient with the CFH1-21::CFHR15-6 hybrid gene we also identified the FH R1210C LPV, which was previously reported as a predisposing factor to a range of pathologies, including aHUS, C3G and AMD. The FH-1210C mutant forms covalently linked complexes with human serum albumin that interfere with FH binding to surface-bound C3b (43, 44). Thus, due to the combination of the FH 1210C mutant from one allele and the hybrid FH with the 1191L and 1197A changes from the other allele, all FH molecules in this patient have a dysfunctional C-terminus, leading to defective regulation of complement on cellular surfaces. Finally, in a third patient carrying the CFH1-21::CFHR15-6hybrid gene, familial studies revealed that the proband also has a de novo duplication involving CFHR3 and CFHR1 genes that was not found in unaffected relatives carrying the hybrid gene.
We found additional genetic or acquired abnormalities even more frequently in the group of patients with SVs involving only CFHR genes, which mostly result in the formation of a CFHR31-5::CFHR410 hybrid. Indeed, 4 out of 6 patients in this group also carry either LPVs or anti-FH antibodies, which would indicate a lower pathogenic impact of CFHR SVs versus those involving CFH. The CFHR31-5::CFHR410 hybrid has already been reported in association with C3G, and it has been suggested that it binds cell surface ligands and favors C3 convertase activity, but functional studies are required to clarify its pathogenic impact (17). The finding that a proband shared the CFHR31-5::CFHR410 hybrid gene, a C3 LPV and CFH H3 risk haplotype with their unaffected mother, underlines the complexity of aHUS, which may not manifest even in subjects with multiple genetic risk factors. It is likely that in such susceptible individuals, environmental factors or an underlying condition that activates complement or perturbs the endothelium are required to trigger the disease. This possibility is in line with the report here of two cases of aHUS secondary to chronic severe hypertension in patients who carried SVs affecting the CFH or CFHRs genes, respectively. It is known that microvascular endothelium can move to a pro-thrombotic phenotype during stress stimuli due to hypertension (45, 46). So in the above patients, the concomitance of hypertension-mediated endothelial stress injury and genetically-determined defective regulation of the complement system may irreversibly compromise the homeostatic equilibrium of the endothelium.
Our data also show relevant associations between the specific SV and disease phenotype, response to therapies and risk of recurrence after kidney transplant. Thus, CFH::CFHR1 hybrid genes were commonly found in patients who manifested the disease in their first year of life, a finding consistent with previously published data (33). The FH::FHR-1 hybrids mimic the effect of LPV in the FH C-terminus that have been often associated with disease onset in infancy (2). Finding that in the majority of CFH::CFHR1 carriers aHUS was triggered by infections, would suggest that in these patients dysfunctional FH could not adequately control the complement activation induced upon the first exposure to viruses or bacteria. As previously reported (37), the reverse CFHR1::CFH hybrid genes were associated with a later disease onset, mostly in adulthood. It is tempting to speculate that in these patients FH produced by the 2 normal copies of CFH, could partially counteract the competitive action of the reverse FHR-1::FH protein (27, 37).
Nonetheless, both patients with hybrid C
留言 (0)