In this study, we reported a potential protein complex consisting of Sema7a (SEMA7A), integrin β1 and NF-κB p105. In this complex, integrin β1 likely acts as a bridge that links Sema7a (SEMA7A) and NF-κB p105 and mediates NF-κB p105 procession and downstream signalling activation, subsequently promoting hepatic inflammation. This pathological process is promoted by Sema7aR145W (SEMA7AR148W) mutation. Meanwhile, Sema7aWT (SEMA7AWT) expression was significantly increased in the HCC patients and mouse model, and might be an important target in clinical preventive treatment measures for alleviating inflammation in tumorigenesis and development.
In previous studies, Sema7aR145W (SEMA7AR148W) reduced the expression of canalicular membrane bile acid transporters, resulting in intrahepatic cholestasis in mice [8]. Additionally, the cell membrane localization of Sema7AR145W and its interaction with integrin β1 were significantly increased in Sema7AR145W mutant mice. Sema7aR145W (SEMA7AR148W) binds to integrin β1 to induce the phosphorylation of PKCα and activation of downstream signals, contributing to intrahepatic lipid accumulation and aggravating NAFLD [9]. Moreover, in this study, Sema7AR145W bound to integrin β1 and activated NF-κB p50/p65 signalling, promoting inflammatory responses. Therefore, according to the definition of gene mutation [38] and the above experimental results, the Sema7AR145W (SEMA7AR148W) mutation could be considered a gain-of-function mutation. However, researches on the regulatory mechanisms of the signalling pathway associated with the Sema7AR145W (SEMA7AR148W) mutation is extremely limited. Therefore, it is necessary to determine the molecular mechanisms involved in the Sema7AR145W (SEMA7AR148W) mutation.
The endogenous and exogenous co-IP data (Fig. 3B, D-G) revealed that integrin β1 could bind to the C-terminus of NF-κB p105. This interaction was elevated in Sema7aR145W homozygous mouse livers. Unfortunately, we could not determine the exact binding site between integrin β1 and NF-κB p105. In addition, since we did not observe the binding of the 50 kDa protein in Fig. 3B, we hypothesized that integrin β1 cannot bind to NF-κB p50 and thus did not verify the interaction between integrin β1 and the N-terminus of NF-κB p105. However, this issue should be demonstrated clearly in our future studies. Meanwhile, because exogenous co-IP can only support the evidence of interactions between two proteins, whether directly or indirectly, we could only confirm that NF-κB p105 was included in the immunoprecipitated proteins of Sema7AR145W (Fig. 3H) and that this interaction between Sema7AR145W and NF-κB p105 was following integrin β1 inhibition. Thus, although we reported that there might exist a new protein complex, the present experimental results only support the information that integrin β1 can directly bind to the C-terminus of NF-κB p105 and probably plays a linking role between Sema7AR145W and NF-κB p105.
Integrin β1 is a receptor of Sema7a (SEMA7A). Sema7a (SEMA7A) interacts with integrin β1, participates in various immunoinflammatory responses, and contributes to proinflammation cytokine release [39,40,41]. In this study, we found that elevated NF-κB p105 procession, advanced NF-κB p50/p65 signalling activation, proinflammatory cytokine production and inflammatory infiltration were displayed in homozygous Sema7aR145W mutant mice. Inhibition of integrin β1 restrained NF-κB p50 generation, subsequently decreasing phosphorylated NF-κB p50/p65 levels and suppressing TNFα and IL1β production (Fig. 3I&J). According to the above results, we propose the hypothesis that integrin β1 acts as a bridge connecting Sema7aR145W and NF-κB p105 and participates in proinflammatory responses in Sema7aR145W mutant mice. Enhanced NF-κB p50 levels largely indicate NF-κB p105 procession. However, we noticed that there was no significant difference in the precursor protein p105 in WT and homozygous Sema7aR145W mutant mice (Fig. 2A&B). Thus, we further evaluated the mRNA level of NFKB1 and found that NFKB1 significantly increased. The increased mRNA level reminded us that there might exist a quick dynamic balance of p105 protein translation and processing (Fig. 2F&G). However, this hypothesis still needs experimental supports, and we would like to focus on this topic in our future studies.
Integrin β1 is a transmembrane protein that mainly mediates cell-to-cell communication in various immunoinflammatory responses. Depending on different microenvironments, different cytokines or chemokines are secreted by different cells. According to these diverse signals, integrin β1 plays a dual proinflammatory and anti-inflammatory role. Thus, the immunoinflammatory consequences of integrin β1 could differ. For example, integrin β1-enriched extracellular vesicles from hepatocytes could mediate monocyte adhesion and contribute to liver inflammation in non-alcoholic steatohepatitis [42]. However, pancreas-specific ablation of integrin β1 is related to widespread inflammation and collagen deposition [43]. In addition, the effects of losing integrin β1 in the same mouse model could also be opposite at different ages [44]. Thus, depending on different microenvironments in different organs and cell types, the proinflammatory and anti-inflammatory functions of integrin β1 could be different but not conflicting. In our study, integrin β1 contributed to NF-κB p50/p65 activation and TNF-α and IL-1β secretion, which play key roles in proinflammation. However, other molecular mechanisms of integrin β1 in the Sema7aR145W (SEMA7AR148W) mutation and high Sema7aWT (SEMA7AR148W) expression in immunoinflammatory responses still need to be characterized in future studies.
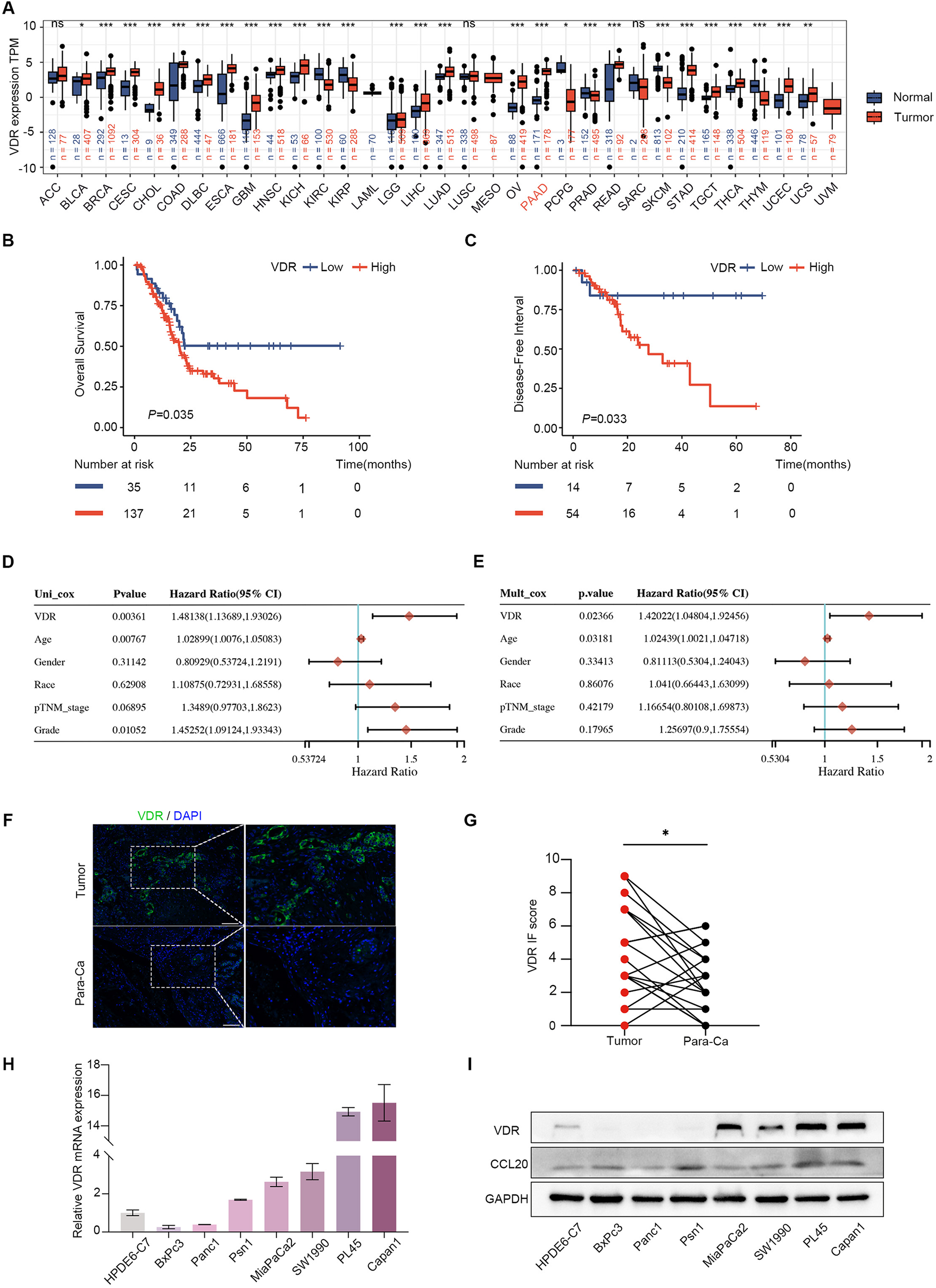
It is well known that HCC, the foremost form of primary liver cancer, is frequently linked with continuous liver inflammation [17, 45, 46]. In a retrospective cohort study of 417 cancer-free patients with cirrhosis, 27% developed liver cancer after approximately 12 years [47]. To determine whether there was relevance between the SEMA7AR148W mutation and HCC, we analysed 1804 HCC patients from the ICGC database. The SEMA7A gene mutation frequency varies widely in different countries; the cohort from China displayed the highest mutation frequency (5.4%) in HCC patients. However, the SEMA7AR148W mutation was not detected in hepatocellular carcinoma but in endometrioid carcinoma (n = 1), adenocarcinoma (n = 2), and oesophageal adenocarcinoma (n = 1). Although the clinical significance of SEMA7AR148W in HCC is limited, its generality of physiological function in gain-of-function mutation and high expression is worth exploring. We noticed that Sema7aWT (SEMA7AWT) was significantly increased in HCC patients and a mouse model (Fig. 5A-C and F–H). Additionally, the interactions between integrin β1 and NF-κB p105 were elevated in the liver cancer group (Fig. 5I). Interestingly, the relevance of Sema7aWT (SEMA7AWT) and HCC has not been previously reported. Further mechanistic studies demonstrated that similar to the Sema7aR145W mutation, NF-κB p50 generation, NF-κB p50/p65 signalling activation, and proinflammation cytokine production were also observed in the HCC group (Fig. 5F-H). In addition, these effects were inhibited by integrin β1 silencing (Fig. 6D-E). Overall, our data indicated that the Sema7AR145W (SEMA7AR148W) gain-of-function mutation and high Sema7AWT (SEMA7AWT) expression had similar effects on inflammation promotion.
Since inflammation is a potential risk factor for tumour occurrence and development, SEMA7AWT is probably an important factor in tumour progression. In addition, we noticed that cell migration and proliferation were stimulated in transfected HepG2 cells. However, some studies have reported that an adequate immune response might be a protective factor in certain cancers, contrasting evidence for protumorigenic functions for inflammation [34]. The molecular mechanisms of SEMA7AWT and its receptors in tumour formation and development need to be determined in our next study.
Based on research on the Sema7AR145W (SEMA7AR148W) mutation [9], the membrane localization of SEMA7A and integrin β1 was thought to increase without expression level changes. Here, we show a new protein‒protein interaction among Sema7a, integrin β1 and NF-κB p105. This interaction strongly increased after Sema7aR145W mutation and promoted the inflammatory response. According to our assumption, this phenomenon might be explained by the upregulated membrane localization of Sema7AR145W (SEMA7AR148W) and integrin β1 benefiting the exposure of their protein structure, especially the binding site for NF-κB p105. Additionally, this protein‒protein interaction and proinflammatory effect still needs to be described in liver nonparenchymal cells and immune cells. These questions need to be answered in our further studies.
In conclusion, we emphasized the important role of Sema7a (SEMA7A) and its receptor integrin β1 in NF-κB p105 procession and NF-κB p50/p65 pathway activation, which had never been reported. Our data also supported that the Sema7AR145W (SEMA7AR148W) mutation and high Sema7aWT (SEMA7AWT) expression both result in NF-κB p50 generation, NF-κB p50/p65 signalling activation and inflammatory cytokine production in an integrin β1-dependent manner.

留言 (0)