
記住我




Sex dimorphisms in coagulation have long been established, with females demonstrating a relative hypercoagulability compared with their male counterparts.1–3 This manifests as a more rapid clot formation, a greater rate of clot propagation, increased functional fibrinogen, increased clot strength, and decreased clot breakdown.4 This sex-specific hypercoagulability translates to clinical significance, with female sex conferring a survival benefit in the setting of trauma-induced coagulopathy (TIC) after severe injury.5 The mechanism driving these dimorphisms and population-level observation of a survival benefit has yet to be elucidated.
Comparative thrombelastographic analysis by sex reveals dimorphisms in several aspects of hemostatic capacity and fibrinolysis; specifically, at baseline, females have shorter time to clot formation, greater rate of clot development, increased clot strength, higher functional fibrinogen, and more activatable platelets than their male counterparts.4 Given the multifaceted dimorphisms in coagulation, the mechanistic drivers in female-specific hypercoagulability are likely multiple and interrelated. A myriad of work has suggested that sex hormones, specifically estrogen, may be driving the coagulation phenotype of females, related to estrogen's action on nitric oxide, the enzymatic coagulation cascade, and plasma cytokines.6–8 Previous work9 has described that not only are there intrinsic sex-based differences in the cellular biology, with platelets demonstrating a sex-specific activation and aggregation potential, but that this can also be manipulated with sex hormones, such that treatment of a male platelet with estradiol “feminizes” the platelet behavior to approximate that of female platelets.
These aforementioned findings highlight that the mechanisms behind sex dimorphisms in coagulation may span both the cellular, enzymatic, and hormonal aspects of coagulation. The objective of this study was to examine the effect of estradiol on clot viscoelastic, thrombin formation, and fibrin biology. We hypothesize that estradiol provokes a hypercoagulable profile in vitro and in vivo, alters the clot proteomic profile, and changes clot structure and fibrin crosslinking.
METHODS Effect of Estradiol on Hemostatic Capacity In VitroHealthy adult (≥18 years) volunteers were recruited from a single, urban hospital and screened via a questionnaire (Supplemental Data Content [SDC] 1, https://links.lww.com/TA/C743) to determine eligibility. This study was approved by the local institutional review board (IRB 14-0366), and all subjects provided informed consent. Subjects were excluded if they had any acute or chronic health problems or were taking medication known to affect coagulation. Females were excluded if they were taking hormonal birth control or any form of hormonal therapy; additionally, the questionnaire screened for and excluded those taking anticoagulant or antiplatelet medications, those with an overweight or obese body mass index, and those who used tobacco products.
Whole blood was collected in citrated tubes (Greiner Bio-One, Monroe, NC; for citrated native and functional fibrinogen thrombelastography), heparin tubes (Greiner Bio-One; for platelet mapping thrombelastography), and sodium citrate-corn trypsin inhibitor (CTI) vacuum tubes (3.2% sodium citrate with 100 μg/mL CTI; for whole blood thrombin generation [TG][) from 15 premenopausal females (average age, 29 years, range, 27–34 years) and 15 years age-matched males (average age, 28 years; range, 27–33 years).
To examine the effect of estradiol on clot viscoelastics and thrombin formation, citrated native (CN-) thrombelastography (TEG), functional fibrinogen TEG (CFF-TEG), and platelet mapping (TEG-PM) were performed after incubation of the blood with physiologic concentrations (final concentration 102 pg/μL) of beta-estradiol (Sigma Aldrich Co.; dissolved in minimal quantity of 0.9% NaCl) for 15 min at 37°C (control without estradiol addition). Citrated native TEG was performed so as to observe clot formation in an unmanipulated fashion, as the additives in activated TEGs used in the clinical setting may affect the subtleties of hemostatic capacity. The TEG assays were performed using the TEG 5000® Thrombelastography Hemostasis Analyzer (Haemonetics Inc., Niles, IL) as described by manufacturer guidelines. Through glycoprotein IIb/IIIa receptor inhibition with abciximab, the citrated functional fibrinogen (CFF-) TEG estimates the relative contribution of “functional” fibrinogen to clot strength through antagonism of platelet contribution to clot strength. Thrombelastography PM quantifies platelet adenosine diphosphate (ADP) and thromboxane A2 receptors' contribution to fibrin clot formation by eliminating thrombin activity with reptilase and Factor XIII in heparinized blood. Citrated native TEG yields the following variables: reaction time (R, time to clot formation), k time (interval from R to fibrin crosslinking providing enough clot resistance to produce a 20-mm amplitude), angle (rate of clot propagation), maximum amplitude (MA, maximal clot strength) and percent clot lysis 30 minutes after reaching MA (LY30). Citrated functional fibrinogen TEG yields the functional fibrinogen level (FLEV) or an estimate of fibrinogen's contribution to overall clot strength. Thrombelastography PM measures the percent platelet inhibition in presence of ADP or arachidonic acid (AA).
In addition, whole blood TG was performed after pretreatment of the blood with physiologic concentrations of beta-estradiol (as described above) with the Stago WB TG device (Near Patient Testing TG [NPT-TG]). The device, which has been described and validated in previous studies,10–12 functions by activating TG by relipidated tissue factor, after which point, the reaction is continuously monitored by means of a thrombin-specific fluorogenic substrate; change in fluorescence intensity produced by the cleavage of the fluorogenic substrate by thrombin is monitored over time and compared with an internal calibrator with known stable thrombin activity. Whole blood TG was initiated using 6.5 pM relipidated tissue factor reagent and 15 mM CaCl2 (Stago, US), and TG curves were recorded continuously for 90 minutes at a rate of three readings per minute alongside an internal calibrator. Whole blood TG yields the following measurements: lag time (time to initial thrombin formation in minutes), peak thrombin (maximum thrombin produced in nM), and maximum rate of TG (velocity index, measured in nM/min).
Effect of Estradiol on Hemostatic Capacity In VivoTo correlate any changes observed in vitro from sex hormone addition, we also measured changes in hemostatic capacity through the menstrual cycle to correlate in vivo circulating sex hormone levels with changes in coagulation. This was accomplished through a longitudinal prospective cohort study of healthy adult women ages 18–45 with a standard 28-day menstrual cycle and without hormonal birth control as a home medication. After three months of menstrual cycle logging (to assure cycle regularity) and a negative pregnancy test, 25 women were enrolled. Whole blood was drawn (3.2% sodium citrate, Greiner Bio-One, Monroe, North Carolina) at peak estradiol levels during the mid-luteal phase (Day 20 of menstrual cycle) and lowest estradiol levels (Day 1 of menses) according to the length of their menstrual cycle.13 CN-TEG was performed as described above.
Effect of Estradiol on Proteomic Profile and Fibrin-Cross LinkingTo also assess the changes in plasma milieu provoked by estradiol, we directed attention to the proteomic composition of the clot and the FXIII-generated crosslinks within the fibrin clots. Blood samples were processed using previously published methods.14 Briefly, whole blood was collected from aforementioned healthy volunteers and two 300-μL aliquots were placed in Eppendorf tubes. One aliquot was treated with estradiol (estradiol added before the addition of TF, MgCl, and CaCl), and then whole blood clots were formed in vitro over a period of 30 minutes. These were washed with Gnd-HCl to isolate the insoluble fibrin matrix, chemically and enzymatically digested, and then fractionated with high pH reversed phase (HPRP) chromatography. Samples were analyzed by nano-UHPLC-MS/MS (Easy-nLC1200, Orbirap Fusion Lumos Tribrid, Thermo Fisher Scientific). For global proteomics, samples were searched using ProteomeDiscoverer (Version 2.1, ThermoFisher). For crosslink analysis, samples were searched using pLink (v2.3.0).
Effect of Estradiol on Clot StructureTo examine the effects of estradiol on fibrin clot architecture, we measured the polymerization of Alexa-488 labeled fibrinogen (Thermo Fisher Scientific, Waltham, MA) using a modification of previously published methods.15 For confocal microscopy, 1.0 mL frozen plasma samples from a stock of pooled donor plasma was thawed in a 37°C water bath with 20 μL of CTI. 4.66 μL of Alexa-488 labeled fibrinogen, 3 μL 0.5 M CaCl2 solution, 5 μL β-estradiol solution (Sigma-Aldrich, St. Louis, MO) and 3.6 μL tissue factor solution (Diagnostica Stago, Parsippany, NJ) with phosphatidylcholine-phosphatidylserine were added to 88.8 μL of thawed plasma, pipetted onto glass bottom microwell dishes (MatTek Corporation, Ashland, MA) and allowed to clot for 20 minutes. Beta estradiol solutions were undiluted 60,000 pg/mL stock, or diluted from stock to achieve final concentrations of 1500 pg/mL, 300 pg/mL, and 60 pg/mL. Hepes-buffered saline was used as a control. Formed clots were fixed with HistoChoice (VWR Chemicals, Solon, OH) and treated with SlowFade Diamond Antifade agent (Invitrogen, Carlsbad, CA). Three-dimensional 20 micron thick “Z-stack” images series were acquired at 0.125 micron steps using a Nikon A1R confocal microscope (Nikon Corporation, Tokyo, Japan) at 60 × 2.0 magnification. Samples were replicated across multiple days (nine replicates of each condition, 45 clots total) to ensure consistent sample preparation, fluorophore performance, and microscope performance.
Using this methodology, we evaluated macroscopic fibrin structure using custom in-house software to provide a quantifiable measurement of fiber resolvability that is relevant to hemostatic potential, as previously described.16 In brief, “fiber resolvability index” is a measurement of the distinctness or clarity of fibrin polymerization. Analysis of fiber resolvability, as determined by standard deviation mapping, provides a reliable assessment of the clot architecture that is unaffected by the overall brightness of the fluorescence. The use of standard deviation as a measure of structural resolvability is a modification of common signal-to-noise or contrast-to-noise ratios and provides an estimate of how much fluorescent fibers are increasing the contrast range throughout the image, regardless of the absolute intensity of the background or the absolute intensity of the fiber fluorescence.
For this and all aforementioned assays, Wilcoxon matched-pairs signed rank test were used to compare control to estradiol-treated samples. R statistical software was used, and significance was defined as p < 0.05.
RESULTS Effect of Estradiol on Hemostatic Capacity In VitroEstradiol provoked a hypercoagulable phenotype on thrombelastographic analysis which was sex-specific. In females, estradiol shortened time to clot formation (median R time, 8.3 min; interquartile range [IQR], 7.1–9.8 vs 11.8 min; IQR 9.3–12.2; p = 0.003), increased the rate of clot propagation (angle 58.6°; IQR, 54.1–62.3° vs 51.7°; IQR, 54.1–62.3°; p = 0.02) and functional fibrinogen (FLEV, 529.2; IQR, 497.7–565.7 vs 392.3; IQR, 350.7–434.9; p = 0.007), increased clot strength (MA, 65.0 mm; IQR, 58.0–71.0 vs 62.0 mm; IQR, 58.5–63.5; p = 0.04), and diminished clot lysis (LY30, 0.8%; IQR 0.2–2.5 vs 2.3%, IQR 1.3–3.4; p = 0.01). In contrast, in males, estradiol did not affect time to clot formation, rate of clot propagation, clot strength, or fibrinolysis (Table 1). Despite no significant changes observed in MA in males with estradiol addition, estradiol affected platelet reactivity in females (ADP inhibition of 99.6% [IQR, 94.7–100.0] vs 54.9% [IQR, 48.2–66.5]; p = 0.003 and AA inhibition of 99.6% [IQR, 81.8–100.0%] vs 67.0% [IQR, 44.9–70.2%]; p = 0.003) and males (ADP inhibition of 83.6% [IQR, 69.4–87.2%] vs 59.0% [IQR, 51.0–72.8%]; p = 0.007 and AA inhibition of 80.2% [IQR, 76.7–86.6%] vs 56.8% [IQR, 51.4–63.8%]; p = 0.003).
TABLE 1 - Changes in Hemostatic Capacity After Estradiol Treatment as Measured by Thrombelastography Control + Estradiol p Females (n = 15) CN-TEG reaction time (min) 10.7 (8.8–12.2) 8.2 (7.2–9.6) 0.0001 CN-TEG k time (min) 3.0 (2.4–3.4) 2.6 (2.1–3.2) 0.10 CN-TEG angle (°) 51.7 (50.0–56.2) 58.6 (54.1–62.3) 0.02 CN-TEG maximum amplitude (mm) 62.0 (58.5–63.5) 65.0 (58.0–71.0) 0.04 CN-TEG LY30 (%) 2.3 (1.3–3.4) 0.8 (0.2–2.5) 0.01 CFF-TEG FLEV 392.3 (350.7–434.9) 529.2 (497.7–565.7) 0.007 PM-TEG ADP inhibition (%) 54.9 (48.2–66.5) 99.6 (94.7–100.0) 0.003 PM-TEG AA inhibition (%) 67.0 (44.9–70.2) 99.6 (81.8–100.0) 0.003 Males (n = 15) CN-TEG reaction time (min) 10.9 (9.3–12.0) 9.6 (7.1–11.6) 0.13 CN-TEG k time (min) 3.8 (3.3–4.2) 3.2 (2.1–4.7) 0.56 CN-TEG angle (°) 45.9 (40.0–52.5) 49.9 (42.1–59.9) 0.27 CN-TEG maximum amplitude (mm) 58.5 (44.0–60.2) 55.2 (48.1–61.9) 0.92 CN-TEG LY30 (%) 0.6 (0.4–2.2) 0.8 (0.2–1.9) 0.51 CFF-TEG FLEV 319.3 (292.0–374.1) 346.7 (301.5–401.5) 0.27 PM-TEG ADP inhibition (%) 59.0 (51.0–72.8) 83.6 (69.4–87.2) 0.007 PM-TEG AA inhibition (%) 56.8 (51.4–63.8) 80.2 (76.7–86.6) 0.003Values reported as median and interquartile range.
Upon examination of whole blood TG, there were no difference in TG at baseline between males and females, including lag time (6.4 minutes [IQR, 5.9–7.0 minutes] in males vs 8.3 minutes [IQR, 6.7–12.9 minutes] in females, p = 0.06), peak thrombin (97.2 nM [IQR, 85.4–131.1 nM] in males vs 79.2 nM [IQR, 43.2–96.9 nM] in females; p = 0.10), and maximum rate of TG (29.6 nM/min [IQR, 22.4–37.4 nM/min] in males vs 20.6 nM/min [IQR, 7.9–24.8 nM/min] in females; p = 0.12). However, with addition estradiol in vitro, similar results were seen as reaction time on thrombelastography. Estradiol provoked a shortening of lag time (6.3 minutes [IQR, 5.8–7.0 minutes] vs 8.3 minutes [IQR, 6.7–12.9 IQR minutes], an increase in peak thrombin (96.6 nM [IQR, 81.6–124.7 nM] vs 79.2 nM [IQR, 43.3–96.9 nM]), and an acceleration of the rate of TG (30.3 nM/min [IQR, 19.6–39.4 nM/min] vs 20.6 nM/min [IQR, 7.9–24.8 nM/min]) (p < 0.0001 for all) in females, with no changes in males (Table 2).
TABLE 2 - Changes in Whole Blood TG After Estradiol Treatment Control + Estradiol p Females (n = 15) Lag time (min) 8.3 (6.7–12.9) 6.3 (5.8–7.1) 0.007 Peak thrombin (nM) 79.2 (43.3–96.9) 96.6 (81.6–124.7) 0.008 Maximum rate of TG (nM/min) 20.6 (7.9–24.8) 30.4 (19.6–39.5) 0.008 Males (n = 15) Lag time (min) 6.4 (5.9–7.0) 6.8 (6.0–7.5) 0.99 Peak thrombin (nM) 97.2 (85.4–131.1) 96.0 (82.7–111.3) 0.31 Maximum rate of TG (nM/min) 29.6 (22.4–37.4) 24.9 (21.3–35.4) 0.44Values reported as median and interquartile range.
Corresponding changes seen in clot formation time and clot strength with estradiol were observed in vivo (Table 3). Specifically, k time was significantly shorter on Day 20 on peak estrogen state (2.3 minutes [IQR, 1.8–2.5 minutes] vs 2.5 minutes [IQR, 2.3–3.1 minutes], p = 0.01). Similarly, the MA was found to be significantly higher on Day 20 as compared with Day 0 (70.0 mm [IQR, 56.5–72.0 mm] vs 59.0 mm [IQR, 58.0–62.0 mm], p = 0.005). There were no statistically significant differences seen in angle or LY30 (Table 2).
TABLE 3 - Changes in Hemostatic Capacity in Estrogen Nadir and Peak of the Menstrual Cycle (n = 25) Day 0 (Estrogen Nadir) Day 20 (Peak Estrogen) p Reaction time (min) 10.2 (9.2–12.0) 11 (10.0–12.6) 0.34 k time (min) 2.4 (2.3–3.1) 2.4 (1.8–2.5) 0.01 Angle (°) 56.5 (51.4–58.4) 56.6 (51.3–57.4) 0.66 Maximum amplitude (mm) 59.0 (58.0–62.0) 70.0 (56.5–72.0) 0.005 LY30 (%) 2.6 (1.3–3.9) 4.1 (1.5–6.7) 0.1Values reported as median and interquartile range.
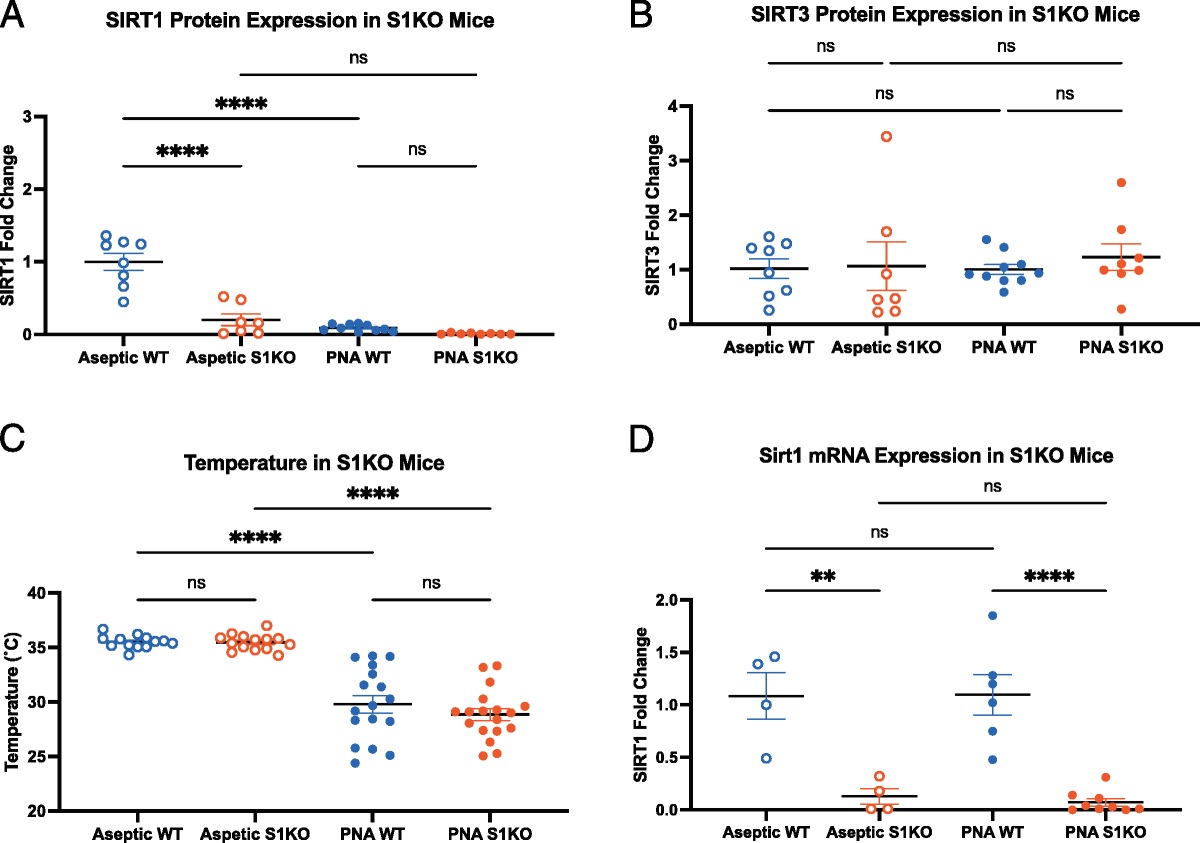
On proteomic analysis, estradiol was associated with robust changes in the proteomic profile. Of the 403 proteins included in the proteomic profile output, estradiol was associated with an increase in 231 in females and 298 in males (Supplemental Digital Content 2, https://links.lww.com/TA/C744). In females, estradiol increased abundance of over 250 proteins including several procoagulant and antifibrinolytic proteins (Table 4). In females, estradiol caused a 5.5-fold increase in apolipoprotein A-I (p = 0.008) and 3.1-fold increase in apolipoprotein A-IV (p = 0.01), 3.0-fold increase in alpha-2-antiplasmin (p = 0.03), 2.1-fold increase in alpha-1-antitrypsin (p = 0.03), 1.6-fold increase in fibrinogen gamma chain (p = 0.02), 1.4-fold increase in fibrinogen beta chain (p = 0.02), and 1.4-fold increase in complement factor H (p = 0.01).
TABLE 4 - Coagulation-Related Proteins Which Are Associated With Increased Abundance After Estradiol Treatment Females Protein Control + Estradiol p Apolipoprotein A-I 160 (37–414) 1215 (329–1892 0.008 Apolipoprotein A-IV 20 (8–86) 87 (34–269) 0.01 Alpha-2-antiplasmin 15 (3–51) 67 (5–149) 0.03 Alpha-1-antitrypsin 628 (170–1060) 108 (59–252) 0.03 Fibrinogen gamma chain 683 (260–1642) 1395 (484–2380) 0.02 Complement factor H 206 (42–232) 36 (18–330) 0.01 Fibrinogen beta chain 360 (50–1530) 750 (179–2020) 0.02 Males Protein Control + Estradiol p Apolipoprotein A-I 21 (8–109) 76 (14–150) 0.03 Plasminogen 74 (26–196) 160 (81–248) 0.01 Vitamin K–dependent protein S 4 (1–14) 19 (5–37) 0.01 Plasma protease C1 inhibitor 16 (2–50) 20 (14–130) 0.04 Histone H1.3 37 (1–271) 89 (15–680) 0.03 Complement C1s subcomponent 1 (0.4–7) 12 (3–21) 0.03 Alpha-2-antiplasmin 5 (0.7–10) 18 (2–33) 0.01Values reported as median area under the curve in million units with interquartile range.
In males, estradiol increased the abundance of over 310 proteins, many of which are related to coagulation as well (Supplemental Digital Content 2, https://links.lww.com/TA/C744). Specifically, estradiol caused a 1.4-fold increase in apolipoprotein A-1 as seen in females (p = 0.03), 1.6-fold increase in plasminogen (p = 0.01), 2.3-fold increase in protein S (p = 0.01), 2.4-fold increase in protease C1 inhibitor (p = 0.04), 2.5-fold increase in histone H1.3 (p = 0.03), 4.1-fold increase in complement C1s subcomponent (p = 0.03), and 4.2-fold increase in alpha-2-antiplasmin (p = 0.01) as seen in females.
Not only were changes seen in the proteomic profile after treatment with estradiol, but crosslinking mass spectrometry analysis revealed addition of estradiol increased the abundance of the fibrin crosslinking itself in both sexes. In females, estradiol caused a significant increase in FXIII crosslinks within the FibA alpha chain of fibrin (median area under the curve [AUC] in million units increased to 1710 [IQR, 18–2870] from 185 [IQR, 25–1310], p = 0.03). Similarly, in males, estradiol also caused a significant increase crosslinks (median AUC in million units increased to 826 [IQR, 26–1,700] from 166 [IQR, 24–481], p = 0.007).
Effect of Estradiol on Clot StructureOn plasma clot architecture analysis, the fiber resolvability increased with estradiol concentration in the mixed donor sex plasma, signifying more highly structured and distinct fibrin fibers (Fig. 1), however this failed to reach statistical significance (median, 160.0 [IQR, 150.0–176.0] vs 130.0 [IQR, 91.5–161.0], p = 0.09).
 Figure 1:
Figure 1: Representative changes in fiber resolvability index after treatment with estradiol.
DISCUSSIONWhile sex dimorphisms in coagulation are well established1–3 and female-specific hypercoagulability has been linked to a survival benefit after injury,5 the mechanisms underlying these phenomena have yet to be fully elucidated. In this multi-tiered experimental design, our data demonstrate estradiol's capacity to provoke hypercoagulability in a sex-specific fashion and manipulate clot biology, ultimately highlighting its mechanistic role in sex dimorphisms in coagulation. In vitro, in females, estradiol shortens time to clot formation, increases TG, increases clot propagation and functional fibrinogen, increases clot strength and platelet reactivity, and decreases fibrinolysis. In vivo, the same effects are observed in time to clot formation and clot strength during estrogen peak of the menstrual cycle. In males, estradiol provokes increased platelet reactivity in vitro. Beyond these enzymatic and cellular effects, estradiol also impacts the clot phenotype itself in both sexes, affecting hundreds of proteins in the proteomic profile which translate to differential fibrin crosslinking. This work contributes to a gap in the current literature on sex dimorphisms in coagulation and has implications for the potential role of estradiol as a therapeutic agent in the treatment of trauma-induced coagulopathy, in particular in female patients.
Our in vitro and in vivo findings highlight the complex and multifold effects of estradiol. The procoagulant effect of estradiol on time to clot formation and whole blood TG in vitro and in vivo supports previous literature which describes a procoagulant profile in women in various versions of a hyperestrogen state, including ovulation induction for in vitro fertilization,17,18 oral contraception,8 hormone therapy for transgender women,19 and pregnancy.20 Further, our in vivo work contributes to previous literature on the dynamic changes in coagulation through a menstrual cycle and estrogen fluctuations; previous reports have described increased fibrinogen and von Willebrand factor antigen and activity and platelet aggregation activity in the peak estrogen phase of the menstrual cycle,21,22 which corresponds with our thrombelastographic analysis. These reported data further contribute to a growing body of literature characterizing sex dimorphisms in platelet biology, with estradiol provoking an increase in clot strength and platelet reactivity on thrombelastography in both sexes. In males, estradiol did not increase MA on CN-TEG, but did increase platelet reactivity on PM-TEG, which may be explained by the fact that the latter assay provides a more detailed quantification of platelet function, including the contribution of ADP and thromboxane A2 receptors to clot formation; in contrast, the maximum amplitude measurement from CN-TEG is a more generalized metric of collective platelet and fibrinogen function.23,24 Previous literature describes greater clotting tendency, reduced response to clopidogrel, more activatable GPIIb-IIIa receptors, and greater posttreatment reactivity on antiplatelet therapy in females,25–27 all of which may be driven directly by the effects of circulating estrogen on estrogen receptors on platelets.28 Beta-estradiol has been linked in increased platelet RNA and release of platelet-derived nitric oxide,29 which may be part of the complex mechanism by which estrogen provokes platelet hyperactivity.
Beyond the effects of enzymatic and cellular hemostatic capacity observed in vitro and in vivo, estradiol also has a drastic impact on the proteomic milieu in which a clot forms in both sexes, with hundreds of proteins in increased predominance after estradiol treatment and subsequent increased abundance of fibrin crosslinks. We suspect these changes in the proteome may indeed be because of platelets and their degranulation products, given the previous discussed work which documents an effect of sex hormones on platelets and their releasates, but we believe the results of this work also suggest that estradiol may be affecting a milieu of proteins, which may expression of and/or interaction of agonists and inhibitors of enzymatic and cellular hemostasis. This contributes to previous work in which plasma proteomic analysis revealed sex-specific profiles, with higher pregnancy zone protein, alpha-1-antitrypsin, beta-2-microglobulin, and complement proteins in females.30
There are several important findings from the proteomic analysis. First, in females, estradiol caused a 1.6-fold increase in fibrinogen gamma chain and 1.4-fold increase in fibrinogen beta chain. Previous work has identified higher levels of fibrinogen in young females, as compared with their male counterparts, by Von Clauss assay.31 Taken together, these data explain why estradiol would also provoke an increase in rate of clot propagation on thrombelastography, as observed in our data. However, the finding of an increased in functional fibrinogen is a novel contribution to our understanding of sex dimorphisms in coagulation, suggesting that even if the fibrinogen levels were the same between sex, the fibrinogen of females has a distinct biology, which may confer rationale for selective transfusion of cryoprecipitate from female donors. Another interesting finding is that in both males and females, estradiol increased apolipoprotein-A. These findings are perplexing since previous work suggests that apolipoprotein-A decreases platelet activation and clot strength32 but our in vitro work described an increase in platelet reactivity and clot strength with estrogen treatment. This highlights the incredible complexity of these interactions, as the hundreds of proteins associated with an increased abundance after estradiol treatment cross-communicate to create a net effect. In addition, estradiol increases multiple complement proteins in both males and females; while the alternative pathway of complement has been linked to hypercoagulability and diminished fibrinolysis,33 the complement system is vastly complex and understanding the sum effect of estradiol on complement function may be hard to predict. Lastly, estradiol was also found to increase alpha-2-antiplasmin in both males and females. Given females' tendency toward a baseline of fibrinolytic shutdown and a decrease seen in LY30 with estradiol treatment in our in vitro work, an increase in alpha-2-antiplasmin is fitting. In males, however, estradiol did not provoke changes in LY30 on the thrombelastographic analysis; it may be that the concomitant increase in plasminogen by estradiol in males combats the effects of the simultaneously increased alpha-2-antiplasmin.
While there is a growing body of literature describing the interaction of estradiol with clot architecture, these data are the first of their kind to describe changes in FXIII crosslinks within FibA, which are significantly increased in the presence of estradiol. These results are in support of previous literature which describes increased fibrin fiber diameter and dense matted clots on scanning electron microscopy in women on combined oral contraceptives8 and more complex and dense fibrin networks during peak estrogen of the menstrual cycle as compared with estrogen nadir.34 While our findings with fluorescent fibrinogen failed to find statistically significant differences, the clot architecture appeared evidently visually different (as displayed in Fig. 1) and perhaps with a larger sample size, our clot resolvability index would have gained significance, as would be suggested by the statistically significant changes observed in the fibrin crosslinks.
The provoked hypercoagulability observed in this work ultimately highlights the potential role for estradiol as a therapeutic agent in the setting of TIC and other forms of coagulopathy. Specifically, the data from this paper support estradiol affecting multiple facets of hemostatic capacity in females and provoking increased platelet reactivity, proteomic profiles associated with coagulation, and fibrin crosslinking in males. As a result, we believe that while the effects of estradiol may not be the same as a therapeutic agent in male versus female patients, we believe that there is potential for benefit in both sexes in its effects of protein and cellular biology. This has been implicated by previous literature which describes unique, organ-specific effects of estradiol after injury.35,36 A myriad of animals models of hemorrhagic shock and trauma have suggested beneficial effects of estradiol therapy, including mitigation of trauma and hemorrhagic shock-related intracellular stress and mitochondrial dysfunction,37 improvement in cardiac function after trauma,38 decreased inflammatory response to injurious insult,39 mitigated secondary injury from traumatic brain injury,40 restoration of hepatocellular function after injury,41 and decreased mortality after trauma.42,43 The mechanistic link between estradiol and improved outcomes after trauma is further strengthened by literature of murine models describing restoration of immune, cardiovascular, and hepatocellular functions function with estradiol supplementation to ovariectomized small animals after trauma hemorrhage.44,45 In addition to the small and large animal-based research related to estrogen and TIC, there is also clinical literature, which links conjugated estrogen therapy with reduced transfusion and coagulation factor requirements, including in orthotopic liver transplantation,46 highlighting the vast therapeutic implications of the provoked hypercoagulability of estrogen described in our work. These implications may span across the estrogen states of females, as it is possible postmenopausal women have preserved functional estradiol-responsive signaling machinery and/or may also have a distinct lineage of platelets after a lifetime exposure of the bone marrow (and megakaryocytes) to estradiol.
Limitations of this work include a small sample size and therefore a higher likelihood of type II error. However, these findings correlate with changes observed in other higher-powered work and the strength of statistical significance is notable. For the in vitro work, the timing of the menstrual cycle for the female donors was not known and as demonstrated by our in vivo work, this may affect our results; however, given the provoked hypercoagulability by estradiol, the timing of the menstrual cycle of a donor may actually underestimate, not overestimate, the effects observed in our data. While females were excluded if they were taking hormonal birth control or any form of hormonal therapy, we did not collect lifelong estrogen exposure history (time of first menses, pregnancy history, etc) in the donors, therefore there may be an underappreciated effect from lifelong estrogen. Lastly, our plasma clot data were based on pooled plasma from a cohort of mixed sexes, and therefore, we may have observed different effects if we were able to stratify those samples by sex.
In conclusion, these data provide a comprehensive depiction of the powerful effects of estradiol on hemostatic capacity and clot biology. This highlights estradiol as one of the mechanistic mediators of sex dimorphisms in coagulation and its wide range of effects, spanning the enzymatic, cellular, and clot structure phenotype of a patient. Ultimately, a mechanistic understanding paves the way for novel therapeutic considerations and pursuits. These data suggest that donor sex should be considered in transfusion practices, and that estradiol may serve as a future potential therapeutic adjunct in resuscitation of coagulopathy.
AUTHORSHIPJ.R.C. performed the literature search, data collection, data analysis, writing, and critical revision. E.E.M., K.H., K.F., M.J.C., and C.C.S. contributed to the study design, data interpretation, writing, and critical revision. N.D. and L.S. contributed to data collection, data analysis, and critical revision.
DISCLOSUREThe authors declare no conflicts of interest.
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health (T32 GM008315 and P50 GM049222). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or other sponsors of the project. This research was also supported with materials from Haemonetics and Stago.
REFERENCES 1. Fourel V, Gabastou JM, Desroys du Roure F, Ehrhardt N, Robert A. Influence of age, sex and ABO blood group on activated partial thromboplastin time. Haemostasis. 1993;23(6):321–326. 2. Roeloffzen WW, Kluin-Nelemans HC, Mulder AB, Veeger NJ, Bosman L, de Wolf JT. In normal controls, both age and gender affect coagulability as measured by thrombelastography. Anesth Analg. 2010;110(4):987–994. 3. Gorton HJ, Warren ER, Simpson NA, Lyons GR, Columb MO. Thromboelastography identifies sex-related differences in coagulation. Anesth Analg. 2000;91(5):1279–1281. 4. Coleman JR, Moore EE, Sauaia A, Samuels JM, Moore HB, Ghasabyan A, et al. Untangling sex dimorphisms in coagulation: initial steps toward precision medicine for trauma resuscitation. Ann Surg. 2020;271(6):e128–e130. 5. Coleman JR, Moore EE, Samuels JM, Cohen MJ, Sauaia A, Sumislawski JJ, et al. Trauma resuscitation considerations: sex matters. J Am Coll Surg. 2019;228(5):760–768. 6. Artero A, Tarin JJ, Cano A. The adverse effects of estrogen and selective estrogen receptor modulators on hemostasis and thrombosis. Semin Thromb Hemost. 2012;38(8):797–807. 7. Frink M, Pape HC, van Griensven M, Krettek C, Chaudry IH, Hildebrand F. Influence of sex and age on mods and cytokines after multiple injuries. Shock. 2007;27(2):151–156. 8. Emmerson O, Bester J, Lindeque BG, Swanepoel AC. The impact of two combined oral contraceptives containing ethinyl estradiol and drospirenone on whole blood clot viscoelasticity and the biophysical and biochemical characteristics of erythrocytes. Microsc Microanal. 2018;24(6):713–728. 9. Coleman JR, Moore EE, Kelher MR, Samuels JM, Cohen MJ, Sauaia A, et al. Female platelets have distinct functional activity compared to male platelets: implications in transfusion practice and treatment of trauma-induced coagulopathy. J Trauma Acute Care Surg. 2019;87(5):1052–1060. 10. Ferrara MJ, MacArthur TA, Butenas S, Mann KG, Immermann JM, Spears GM, et al. Exploring the utility of a novel point-of-care whole blood thrombin generation assay following trauma: a pilot study. Res Pract Thromb Haemost. 2021;5(3):395–402. 11. Dunbar NM, Chandler WL. Thrombin generation in trauma patients. Transfusion. 2009;49(12):2652–2660.
留言 (0)