
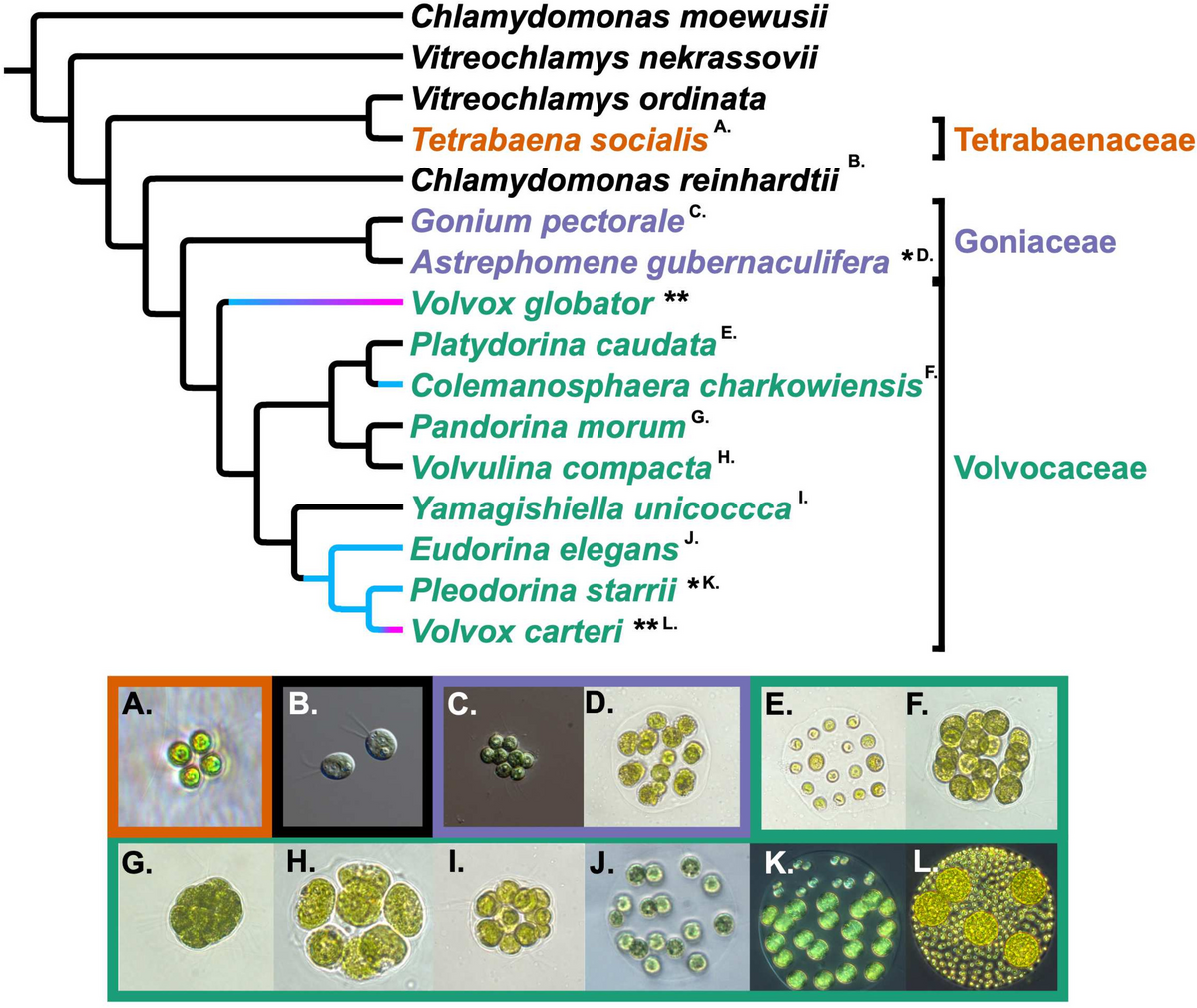
記住我


The feature vector dimensions of two of the five physicochemical descriptors, i.e., QSorder and APAAC, are dependent on the algorithm parameters. To make each type of feature as informative as possible, the related parameters were optimized before they were used for feature optimization. The parameter search range and the optimal value are listed in Additional file 1: Table S2. After the parameter value is determined, the combined feature dimension generated by the five physicochemical-based descriptors is 523D. To reduce the computing complexity and avoid the overfitting issue of the machine learning model, the genetic algorithm was applied to choose the optimal feature subset from the combined features. The number of populations was set to 200, and the chromosome length and the number of generations were set to 100 and 500, respectively (refer to the “Methods” section for details). To evaluate the effectiveness of the genetic algorithm for informative feature identification, four other widely used two-step feature selection strategies were used for comparison. In the first step, four types of feature importance values, calculated by random forest (RF), light gradient boosting machine (LGB) [14], F-score, and MRMD [15], were calculated to yield four descending order lists. In the second step, for each feature list, the optimal feature subsets were selected using the sequential forward search (SFS) method [16]. Finally, the feature subset leading to the model with the highest AUC value is retained as the optimal feature subset.
The results of the above five feature selection strategies are presented in Fig. 1. Of note, the feature dimension of the genetic algorithm is fixed to 100D as the demand of the algorithm structure, while the dimensions of the remaining four feature importance-based SVM models linearly increase with the number of iterations from 1 to 200. Generally, the five metric values are gradually increased, and the maximum scores are obtained at approximately 100 iterations. Specifically, among the five feature selection strategies, the genetic algorithm resulted in the best performance when evaluated by ACC, SN, SP, MCC, and AUC. LGB (importance_type=’gain’) and RF importance-based methods are ranked at the second level, and MRMD- and F-score-based feature selection methods are proven to be the least effective strategies. It can be observed from Fis. 1A–E that the changing trend of the five metrics is not completely synchronized, so the AUC value is used to choose the best feature subset. As shown in Fig. 1E, the maximum AUC reached 0.6949 at the 117th iteration; therefore, the genes, namely, the features, retained in the 117th generation were kept as the optimal feature subset of the five physicochemical descriptors. The feature dimensions corresponding to the maximum AUC of the above five feature optimization methods are shown in Fig. 1F, where the LGB (96D) and genetic algorithm (100D) methods exhibited the lowest dimensions. Considering the model performance and feature dimension, it can be concluded that the genetic algorithm-based feature selection strategy outperformed the other four methods.
Fig. 1
Comparison of different feature selection methods. A–E Metrices value and feature dimensions based on five feature selection strategies. F Feature dimensions of optimal feature subsets based on the metric AUC of the five feature optimization methods. GA: genetic algorithm.
Distributed representation of amino acid fragmentsAll training protein sequences were divided into a k-mer corpus, and each k-mer was embedded into a 100-dimensional feature using word2vec with a skip-gram model. In this process, two critical parameters, namely, the sliding window (length k of the k-mer) and the number of surrounding words (window size w from word2vec), were optimized. The k value was varied from 2 to 6, and the w value was varied from 1 to 7. The ACC values for all possible combinations of k and w are depicted in Additional file 2: Fig. S1. We observed that the ACC values gradually decreased with the value of k, while the w value had less impact on the ACC value. Taken together, the parameters resulting in the maximum ACC value were adopted for the final skip-gram model, which is based on a k-mer length of 3 and a window size of 2.
DeepSoluE modelIn DeepSoluE, prediction features were combined by the 100D features optimized from the five physicochemical feature descriptors using a genetic algorithm, 100D embedded semantic features, and 19D features related to sequence identity and special physicochemical characters (refer to Methods for details). The combined 219D features were fed into the DeepSoluE architecture. To avoid overfitting, an early stopping strategy based on the validation loss is applied when training the LSTM model. Then, the model was validated. Independent test data were used to test the model that showed the best performance on the validation data. As shown in Table 1, the training and validation processes are measured on the metric ACC, and the independent testing results are measured on all the five metrics. For the ten trained models, the maximum ACC was achieved on Model 6 (0.6574), and the minimum ACC was obtained on Model 4 (0.6386). Then, the trained models were evaluated on the independent test dataset. Model 1 resulted in the best MCC (0.2101) and AUC (0.6254), while Model 5 ranked last on MCC (0.1931) and AUC (0.6163). For the sake of convenience and comparison, the average values of the ten models were used to measure the performance of DeepSoluE. Based on that, DeepSoluE achieved an average training ACC of 0.6463 and an average validation ACC of 0.6401. On independent test data, DeepSoluE achieved an average ACC of 0.5885, SN of 0.6108, SP of 0.5661, MCC of 0.1776, and AUC of 0.6197.
Table 1 Individual and ensemble model performance on training and testing dataAs described in methods, each trained model takes 9 of the ten folds of the complete training dataset as input. To give full play to the advantages of ensemble learning, an ensemble method (soft voting, threshold = 0.4) is applied to build an integration model. As shown in Table 1, the integrated model achieved better performance than the individual model, indicating that the ensemble strategy is effective for model performance improvement.
To further assess the efficacy of the LSTM architecture, we compared DeepSoluE with 11 popular traditional machine learning algorithms, including the AdaBoost classifier (ADAB), bagging (BAG), decision tree (DT), k-nearest neighbor (KNN), light gradient boosting machine (LGB), logistic regression (LR), naïve Bayesian (NB), random forest (RF), stochastic gradient descent (SGD), support vector machine (SVM) and extreme gradient boosting (XGB) algorithms. Each of the 11 models is trained on the training dataset and evaluated on the independent test dataset (refer to Additional file 1: Table S3 for model hyperparameter optimization). Figure 2 presents the values of the five metrics, in which DeepSoluE outperformed all the rest of the classifiers in terms of ACC, SP, MCC, and AUC (Additional file 3: Table S4). For metric SN, the highest value is obtained on the NB classifier, followed by the RF and BAG classifiers. DeepSoluE ranked fourth among the 12 models. It is worth noting that DeepSoluE achieved more balanced performance with |SN-SP|=2.65%, while the NB classifier returned |SN-SP|=27.74%, the RF classifier returned |SN-SP|=15.68%, and the BAG classifier returned |SN-SP|=16.45%. As indicated in Eqs. (2) and (3), SN and SP actually constrain each other as they measure a predictor from two different angles [21, 34]. Maintaining a balance between SN and SP is crucial for an accurate model to provide an unbiased prediction. Overall, these results demonstrate that DeepSoluE is significantly superior and more robust than the traditional classifiers.
Fig. 2
Performance comparison of DeepSoluE and 11 conventional machine learning methods
Feature contribution and dependency analysisSHapley Additive exPlanation (SHAP) values [17] were applied to infer informative features of DeepSoluE. First, the top 20 most important features are calculated and depicted by the SHAP summary plot. As shown in Fig. 3A, physicochemical properties critical for protein solubility include protein isoelectric point, gravy, aromaticity, flexibility, instability index, molecular weight, and fraction charge. Protein structure and motifs/patterns that are related to protein solution are composed of an aa turn, an aa helix, and lysine (K), a polar amino acid group (“KPDESNQT”) that is defined by hydrophobicity attribute PONP930101 and amino acids (“MHKFRYW”) that have a larger residual volume according to the definition of normalized van der Waals volume [18].
Fig. 3
Feature contribution and dependency analysis. A The 20 most important features. B Summary plot for SHAP values. For each feature, one point corresponds to a single sample. The SHAP value along the x-axis represents the impact that feature had on the model’s output for that specific sample. Features in the higher position in the plot indicate the more important it is for the model. C–J SHAP dependence plots. These plots show the effect that a single feature has on the model predictions and the interaction effects across features. Each point corresponds to an individual sample, the value along the x-axis corresponds to the feature value, and the color represents the value of the interacting feature
Furthermore, how the feature values affect the model prediction was explored. Figure 3B shows their corresponding summary plots of the top 20 most important features, which illustrates how high and low feature values were related to the model output. For example, high values of isoelectric point are associated with positive impacts on protein solution, while low values have negative impacts. Similar feature value influences are also observed in the other 12 of the top 20 features. Opposite changing trends are observed by the other 7 features; i.e., high values of identity weaken the model behavior, and low values of identity boost model performance (Fig. 3B). In addition, SHAP values of several decisive features, e.g., isoelectric point (−0.4 to 0.6), identity (−1.25 to 0.75), AAS38 (−0.6 to 0.4), vary in a larger range than others, which suggests why they dominate the model’s behavior; the reverse situation explains why the aa helix (−0.15 to 0.1) and norm Waals volume. G3(CTDC) (−0.15 to 0.1) is less important than the others since changes in its value result in less influence on the corresponding SHAP values.
From the biological aspect, the pH of the solution affects the nature and the distribution of the protein’s net charge, and, generally, protein exhibits the least solubility at the isoelectric point [19]. Therefore, proteins with a higher isoelectric point have a net negative or positive charge, and interact with more water, which may partly explain why isoelectric point was the most important feature and high value has a positive impact on protein solubility (Fig. 3AB). Similar, feature aa_turn related to three hydrophilic amino acids (G, N, and S), amino acids that have a larger flexibility present a larger contact area with the solvent, both of them contributed to protein solubility. Protein sequence that enriched with charged amino acids (R, K, D, E) are also beneficial for their solubility. Notably, several opposite situations are observed on our results, for example, higher polar amino acid content (PONP930101) showed a negative impact on protein solubility (Fig. 3B), this implies factors that influence protein solubility is far from clear and further study on this area is necessary.
Finally, SHAP dependence plots were used to provide meaningful insights into interaction effects across features. The dependence plots of the top 20 features are shown in Fig. 3C–J and Additional file 2: Fig. S2. Feature turning points can be visualized; for example, the proposed DeepSoluE takes approximately 0.4 as a turning point for the feature isoelectric point, and the feature values higher than that value contribute to performance boost (Fig. 3C). The turn point of feature identity is approximately 0.5, and values higher that value change SHAP values from negative to positive. For feature interactions, Fig. 3D shows that high identity values (range 0.4 to 1.0) with low AAs_TMHs values (0.1–0.2) have a negative impact on model behavior (SHAP values<0), while low values of AAs_TMHs show little impact. A high feature value of aromaticity (0.0–0.3) with a low feature value of N_in_cytop contributes to accurate model prediction, while a low feature value of N_in_cytop has the opposite effect. Similar feature interaction patterns were observed in two other feature pairs (Fig. 3H and I). More feature interaction patterns can be seen in Fig. 3C–J and Additional file 2: Fig. S2.
Comparison with existing predictorsThe independent testing set of this study is used to evaluate and compare DeepSoluE with 12 previously published tools. The recommended parameters, such as the model decision threshold (T), of each tool are adopted for result evaluation. Table 2 provides details of the comparative analysis results. DeepSoluE exhibited the best performance when evaluated by the metrics ACC and MCC. Although the best SN and SP were achieved by the SWI and DeepSol models, respectively, the prediction results of the two models are seriously biased. SWI achieved an SN of 0.7781; however, the SP of this model was 0.3400, which resulted in |SN −SP|=43.81%. This finding suggests that SWI tends to predict a query protein as soluble. Similarly, DeepSol resulted in |SN −SP|=76.06%, which means that the prediction result of DeepSol is heavily skewed toward insoluble.
Table 2 Performance comparison of DeepSoluE with existing predictors in protein solubility prediction on independent test dataTo further make a reasonable comparison, models that presented a |SN–SP| < 20% were filtered for further analysis. Based on the preconditions, four models, ccSOL omics, SOLpro, SoluProt, and DeepSoluE, are retained. Among them, DeepSoluE shows the greatest value on metrics ACC, TP, TN, SN, SP, and MCC, followed by SoluProt and SOLpro. Notably, while the datasets of DeepSoluE are homology reduced to 25% and the testing set is independent of the training set, other tools’ training sets might have a high sequence overlap with our test set. For example, the DeepSol and SKADE training sets presented a 74% overlap with our testing set, and SoLpro had an overlap of 15.5%. More information on the sequence identity of previous tools is presented in [11]. Of note, the model with high sequence redundancy between its training set and our testing set will benefit from the comparison results, as listed in Table 2. In conclusion, all these results demonstrate that DeepSoluE outperformed the existing prediction algorithms for protein solubility prediction.
留言 (0)