
記住我










Complement activation causes damage to the skin and muscles in myositis through the formation of membrane attack complexes on capillaries and activation products that stimulate inflammation. How genetic diversity of complement contributes to differential susceptibility among human patients with myositis was unknown.
WHAT THIS STUDY ADDSWe deciphered gene copy number (GCN) variations for complement total C4 (C4T), acidic C4A, basic C4B, long genes (C4L) and short genes (C4S) in >1600 patients with IIM and >3500 healthy subjects of European ancestry. Low GCNs of C4T, C4A and C4L strongly correlated with elevated risk of juvenile dermatomyositis, adult-onset dermatomyositis and polymyositis. The presence of HLA-DR3 with deficiencies for C4A or C4B was the predominant genetic factor for inclusion body myositis. Lower plasma protein levels of C4 and C3 were present among patients with IIM with anti-Jo1 and myositis-associated autoantibodies.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICYLow GCN of complement C4 or C4A deficiency are strong risk factors for autoimmunity in IIM. In the presence of myositis autoantibodies, low complement levels could be both a cause (as complement genetic deficiency possibly causes autoimmune disease) and an effect of the disease (through immune-complex-mediated complement consumption). Thus, monitoring depressed levels of complement and elevated activation products would be informative about disease activities or flares.
IntroductionIdiopathic inflammatory myopathies (IIM) are a group of autoimmune diseases characterised by chronic muscle weakness and fatigue.1–3 Pathology in IIM includes the generation of myositis-related autoantibodies and infiltration of leucocytes into muscles and/or the skin leading to inflammation with high levels of muscle enzymes in the circulation.1 Four major subgroups of IIM include juvenile dermatomyositis (JDM),4 adult-onset dermatomyositis (DM), polymyositis (PM) and inclusion body myositis (IBM). Immune-mediated necrotising myositis and anti-synthetase syndrome are recently defined categories.
JDM is the most common form of myositis in children that has a mean age of diagnosis between 7 and 8 years.4 Patients with JDM have similar muscle and skin manifestations as in adult-onset DM but do not have the increased risk of interstitial lung disease (ILD) and malignancy that are more common among adult patients. Specific patterns of rash involving the eyelids, face, shoulders and body areas frequently exposed to sunlight are prevalent among JDM and DM. Muscle weakness is symmetric and proximal to the body axis. In pathognomonic muscle biopsies, there is remarkable complement-mediated destruction of perivascular endothelium leading to perifascicular ischaemia and degeneration of muscle fibres.5–8 However, triggers for complement activation and whether complement genetic diversity is engaged in the breakdown of immune tolerance have not been investigated. PM is more common in women over the age of 30. Patients with PM mainly have muscle weakness and may develop ILD but skin manifestations are infrequent. For IBM, the disease starts insidiously at elderly age and weakness may involve both proximal and distal muscles. PM and IBM both seem to involve primarily cell-mediated autoimmunity.1
The aetiology of IIM is likely multifactorial. Inflamed muscle cells in patients with IIM express human leucocyte antigen (HLA) class I and sometimes class II proteins that present antigens to T cells and provide activation signals. Many patients with IIM have myositis-specific autoantibodies (MSA) and/or myositis-associated autoantibodies (MAA),9 10 which together are termed myositis-related autoantibodies. MAA are also present in other connective tissue diseases. Intriguingly, patients with IIM with the same autoantibodies may present with similar disease patterns and profiles.9 11
Among subjects of European ancestry, the presence of HLA-DRB1*03:01 or HLA-DR3 tends to strongly associate with complement C4A deficiency, the presence of a single short C4B gene and HLA-B*08:01, which is therefore named the ancestral haplotype AH8.1.12–18 Rothwell and colleagues showed that HLA-DRB1*03:01 was one of the strongest risk factors for IIM.19 20
Complement C4 plays essential roles as an anchor protein in the activation of the classical and the mannan-binding lectin pathways for the humoral immunity (figure 1) to defend against infection.21 22 There are four layers of genetic complexity for human C4, which include (1) multiallelic gene copy number (GCN) variations with 2–10 copies of C4 genes present in a diploid genome among different individuals23–25; (2) gene size dichotomy with a long gene and a short gene depending on the integration of the 6.4 kb endogenous retrovirus HERV-K(C4) into intron 9 of long genes26 27; (3) each C4 gene either codes for an acidic C4A or a basic C4B protein, which differ by four specific amino acid residues between positions 1120 and 1125 coded by exon 26: PCPVLD for C4A and LSPVIH for C4B28 29 and (4) both C4A and C4B proteins are polymorphic with differential electrophoretic, serological and functional reactivities (figure 1).25 28 30 Isotype deficiency of C4A has been shown to be strongly associated with increased susceptibility of lupus in multiple racial groups23 24 31 and in an animal model.32
 Figure 1
Figure 1 The complement system with emphasis on the genotypic and phenotypic diversities of C4A and C4B. (A) Activation and regulation of the human complement system. Activation of zymogens and progression of pathways are shown in red; regulations of activated products in green. A positive feedback of amplification is common for all three activation pathways. (B). Genetic locations for constituents of the C3 convertases for classical and alternative pathways. (C) Segmental duplications with one to five modules of the RP-C4-CYP21-TNX (RCCX) in haplotypes at the class III region of the human leucocyte antigen (HLA). (D) Dichotomy of human C4 gene size with the long gene containing endogenous retrovirus HERV-K(C4) in the ninth intron and the short gene without the endogenous retrovirus. (E) Specific polymorphisms leading the isotypic changes for C4A and C4B proteins. (F) Immunofixation experiments showing the quantitative and qualitative diversities of C4A and C4B protein allotypes including deficiencies. (G) The range of polymorphic variants for C4B (left panel) and for C4A (right panel). CNVs, copy number variations.
The role of C4 isotype deficiencies in myositis is understudied. The continuous GCN variations and associated polymorphisms for C4A and C4B pose challenges for accurate data interpretation through whole-exome or whole-genome sequencing and analyses by Immunochip techniques. In a study of 95 white patients with JDM, we showed that C4A deficiency was a strong risk factor for JDM.33 How complement C4 genetic diversity contributes to disease predisposition in different forms of IIM, the development of MSA and/or MAA and the relative roles of HLA-DRB1*03 and C4A deficiency in IIM have yet to be assessed, however.
We leveraged a robust collection of biospecimens and clinical data for patients with IIM recruited by Investigators of the Myositis Genetics Consortium (MYOGEN) from the UK, Sweden, the Czech Republic, Belgium and the USA, plus geographically matched healthy controls to investigate the GCN variations of total C4 (C4T), C4A, C4B, long C4 genes (C4L) and short C4 genes (C4S) in disease susceptibility for IIM and its four major subtypes. The relative roles of HLA-DRB1*03 and C4A deficiency on genetic risk of IIM, and how the C4 GCN variations and complement protein levels correlated with the presence of myositis-related autoantibodies were also examined.
Patients and methodsStudy populationsOur study population included 1644 patients with IIM and 3526 healthy controls (table 1). Patients with IIM fulfilled Bohan and Peter classification criteria for DM, JDM and PM,34 35 and Griggs or European Neuromuscular Center or the UK Medical Research Council criteria for IBM.36–38 Study subjects were recruited with informed consent from northern and central Europe and the USA through the MYOGEN or at the Nationwide Children’s Hospital, were self-reported European ancestry or based on principal component analysis.19 Patients with non-European ancestries were studied but not included in specific genetic analyses. Healthy control subjects did not report to have an autoimmune disease.
Table 1Demographics of study populations
Isolation of genomic DNA, EDTA-plasma and Southern blot analysesFor subjects recruited in Ohio, preparation of genomic DNA from peripheral blood samples, performance of TaqI, PshAI-PvuII restriction fragment length polymorphisms and PmeI pulsed-field gel electrophoresis to elucidate RP-C4-CYP21-TNX (RCCX) modular structures were as described.39
Copy numbers and sizes of C4A and C4B genes by real-time PCRWhen quantities of genomic DNA were limiting, copy numbers of C4 genes were determined by TaqMan-based quantitative real-time PCR with internal control using cosmid DNA with both test and control amplicons. Five independent test amplicons specific for total C4 (C4T), C4A, C4B, long genes and short genes were performed. Verification was achieved when GCN of C4T=GCNs of C4A+C4B and/or GCNs of C4L+C4S.14
Protein concentrations and polymorphic variantsComplement C4 and C3 protein concentrations were measured by single radial immunodiffusion (RID) using EDTA-plasma and an RID kit from the Binding Site (UK). C4A and C4B protein allotypes in plasma samples were resolved by high-voltage agarose gel electrophoresis, followed by immunofixation using antiserum against human C4.40
Genotyping of HLA-DRB1Genotyping for HLA-DRB1 alleles for samples from the USA and Sweden was performed at low resolution using the sequence-specific primer-PCR methods (eg, DR low-resolution kit: Olerup SSP, Saltsjobaden, Sweden).41 42 The HLA-DRB1 genotypes for samples from the UK were deduced from single-nucleotide polymorphisms data using SNP2HLA software.20 43 44 High concordance of imputed data from DNA sequencing and conventional HLA typing techniques was obtained.20
Statistical analysesThis was a cross-sectional, case–control study. Statistical analyses were performed using JMP16 software from SAS. Continuous data between patients and controls were compared by t-tests. The distributions of C4T, C4A, C4B, C4L and C4S GCN groups in patients with IIM or in each IIM subgroup and controls were analysed by χ2 analyses. The GCN groups for each type of C4 genes were segregated dichotomously into low GCN and medium to high GCN groups, and their frequencies compared between case and controls with χ2 analyses to compute ORs and 95% CIs. The low GCN groups were defined as follows: C4T=2+3, C4A=0+1, C4B=0+1, C4L=0+1+2 and C4S=0. A Bonferroni’s correction for a C4 genotype with p<0.01 was considered significant to account for five structural variants being investigated for IIM genetic risk individually. For intragroup comparisons of a specific phenotype with a genotype, a p-value <0.05 was viewed as significant.
ResultsComparisons of GCN variations of complement C4 between IIM and controlsTotal C4The mean GCN and SD of C4T among patients with IIM was 3.50±0.78, compared with 3.83±0.76 in healthy controls (δ=−0.333, p=1.4×10−46, t-test) (table 2). The most prevalent GCN group for C4T in IIM was 3 copies with a frequency of 45.5%, followed by a GCN group of 4 copies with a frequency of 38.2% (figure 2). Patients with 2 copies of C4T comprised 7.1%, and those with 5, 6, 7 and 8 copies comprised a total of 9.2% of all IIM. Categorically, the distributions of C4T GCNs in IIM were substantially different from those in healthy controls, with a p-value of 1.4×10−53 (χ2 analysis). The OR and 95% CI for IIM subjects with two copies of C4T was 2.26 (1.74 to 2.95), p=2.0×10−9, and those with two or three copies (C4T=2+3) had an OR=2.62 (2.32 to 2.95), p=2.8×10−55 (figure 3A; see also online supplemental figure S2, supplementary results). Thus, low GCNs of C4T, that is, C4T=2 and C4T=2+3, had similar magnitude of effects on the genetic risk of IIM.
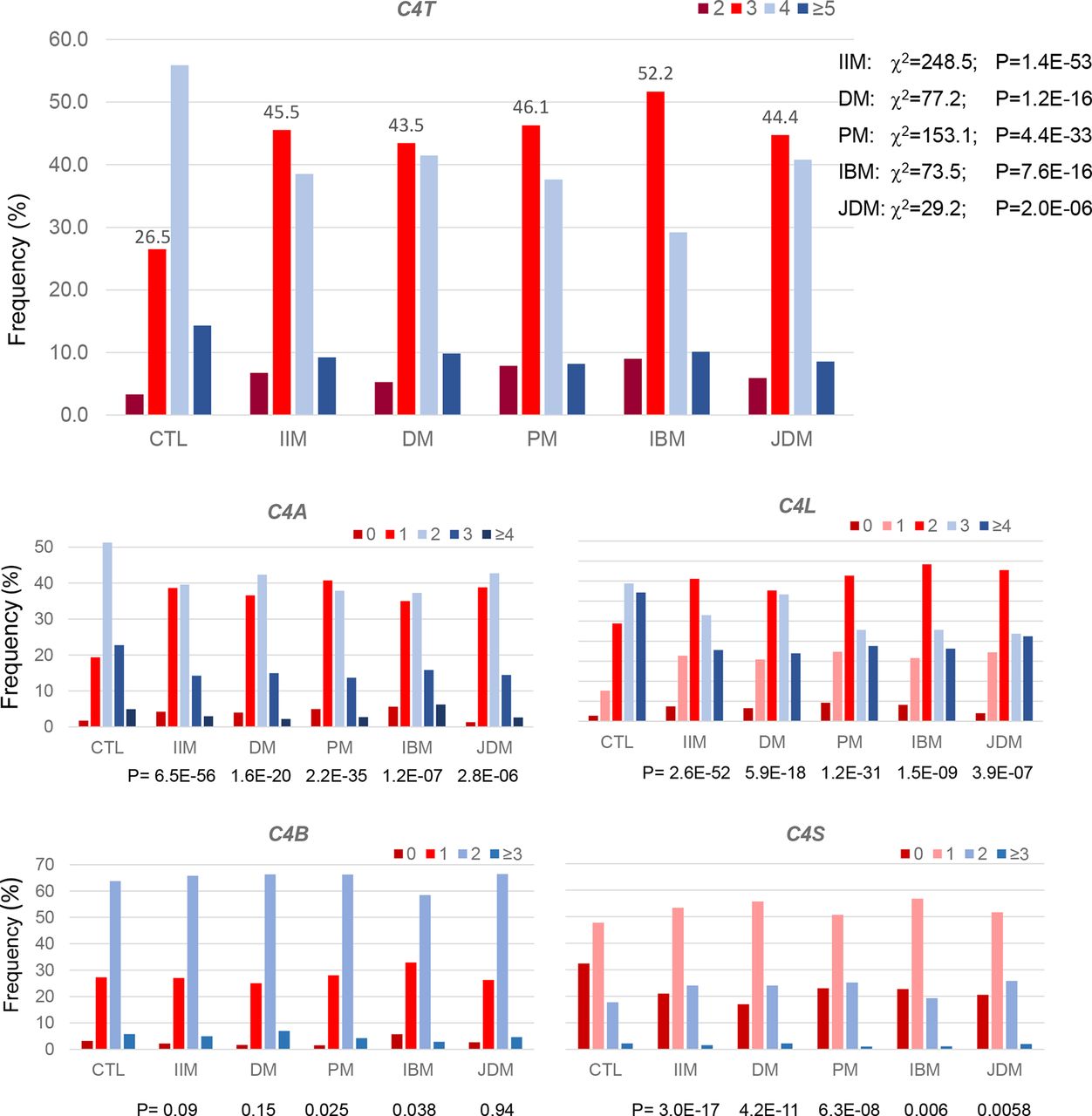 Figure 2
Figure 2 Comparisons in patterns of distributions for complement C4 gene copy number groups for total C4, C4A, C4B, C4L and C4S among healthy controls (CTL) and patients with idiopathic inflammatory myopathies (IIM) including adult dermatomyositis (DM), polymyositis (PM), inclusion body myositis (IBM) and juvenile dermatomyositis (JDM). Frequencies with three copies of total C4 (C4T) were labelled to highlight the difference between patients and CTL. C4A, acidic isotype of complement C4; C4B, basic isotype of complement C4; C4L, long form of C4 gene with human endogenous retrovirus HERV-K(C4); C4S, short form of C4 gene without integration of the retrovirus HERV-K(C4); C4T, total copy number of C4 genes.
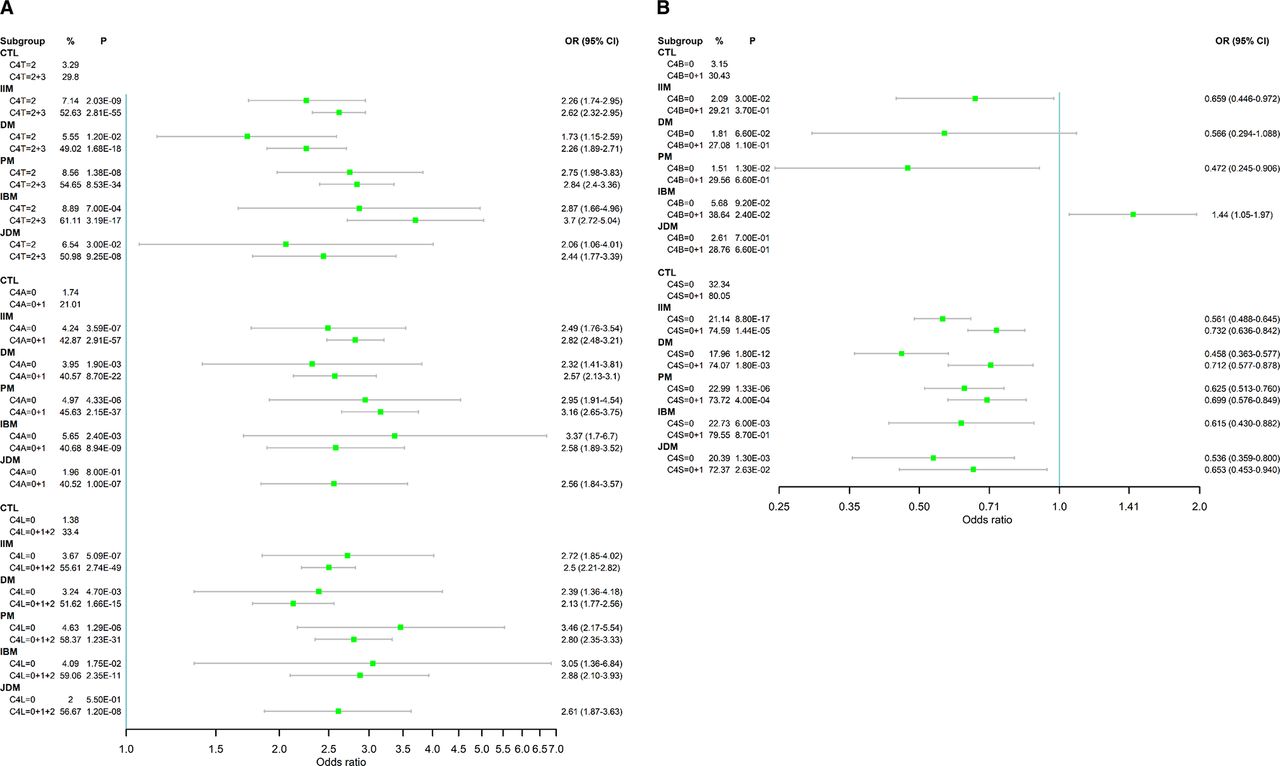 Figure 3
Figure 3 Forest plots of ORs for low copy number groups for C4T, C4A, long genes (C4L) as risk factors (A), and for C4B and short genes (C4S) as protective factors (B) in IIM and subgroups. A single exception was that low copy number C4B was also a risk factor of IBM. Notice the partial dominance of low GCNs of total C4 (C4T=2 and C4T=2+3) and C4A deficiencies (C4A=0 and C4A=0+1) on conferring risk of IIM and its subgroups DM, PM, IBM and JDM. The ORs in panel A are shown in log-scale. C4A, acidic isotype of complement C4; C4B, basic isotype of complement C4; C4L, long form of C4 gene with human endogenous retrovirus HERV-K(C4); C4S, short form of C4 gene without integration of the retrovirus HERV-K(C4); C4T, total copy number of C4 genes; DM, dermatomyositis; GCNs, gene copy numbers; IBM, inclusion body myositis; IIM, idiopathic inflammatory myopathies; JDM, juvenile dermatomyositis; PM, polymyositis.
Table 2Comparisons of mean complement C4 gene copy numbers (GCN) among patients with IIM
C4A in IIMGCN of C4A varied from 0 to 6 among patients with IIM with a mean of 1.74±0.88, compared with 2.10±0.84 in healthy controls (δ=−0.37, p=6.0×10−46). There were remarkable increases in the frequencies of C4A low GCN groups and decreases in medium and high GCN groups in IIM (p=6.5×10−56). While 40.1% of patients with IIM had two copies of C4A genes, those with 0 and 1 copy constituted 4.2% and 38.6% of patients, respectively (figure 2). Patients with 3–6 copies (high GCN) of C4A together had a combined frequency of 17.1%. The OR was 2.49 (1.76–3.54, p=3.6×10−7) for C4A=0 and 2.82 (2.48–3.21, p=2.9×10−57) for C4A=0+1 (figure 3A). The magnitudes of the effects of low C4A GCNs on IIM were similar to that observed in C4T=2 and C4T=2+3.
C4B in IIMUnlike C4T and C4A, C4B copy number group distribution in IIM was almost identical to that observed in healthy controls, which ranged between 0 and 5. Close to two-thirds of the patients with IIM (65.8%) had two copies of C4B, while 2.1% and 27.1% had 0 and 1 copy, respectively. Patients with 3, 4 and 5 copies of C4B constituted a total frequency of 4.9%.
Long genes (C4L) in IIMThe copy number of C4L varied from 0 to 8 in patients with IIM. The mean C4L GCN in IIM was 2.41±1.13, which was significantly lower than that in healthy controls (2.94±1.08, p=1.7×10−54). The distribution of GCN groups for C4L was different from that of controls (p=2.6×10−52). The combined frequency for low GCN of long genes (C4L=0+1+2) in IIM was 55.6%, compared with 33.4% in healthy controls (OR=2.50 (2.21–2.82), p=2.7×10−49) (figure 3A). Decreasing GCNs of C4L elevated the ORs for IIM: 2.55 (2.15–3.03, p=9.4×10−27) for C4L=0+1 and 2.72 (1.85–4.02, p=5.1×10−7) for C4L=0. The frequency of long genes among total C4 decreased from 74.6% in controls to 63.2% in IIM (C4L/C4T, p=2.1×10−53).
Short genes (C4S) in IIMThe copy number of C4S in IIM varied from 0 to 5. The mean copy number was 1.06±0.72, which was higher than that in healthy controls (0.90±0.77, p=3.0×10−12). More than half of the patients with IIM had a single copy of C4S (53.5%). The frequency of subjects lacking C4S (C4S=0) was significantly reduced from 32.3% in controls to 21.1% in IIM (OR=0.56 (0.49–0.65), p=8.8×10−17).
C4 GCN variations among subgroups of IIMCompared with controls, the four IIM subgroups had lower mean GCNs of C4A in the range of 1.70 to 1.82 but they were not distinguishable among themselves (table 2 and figure 2). Patients with IBM were unusual for having lower GCNs of C4B (1.59±0.65) than other IIM subgroups. In the other three subgroups, lower C4T GCN was primarily attributable to the decreased GCN of C4A.
As shown in figure 3, the effect sizes of C4T=2+3, C4A=0+1 and C4L=0+1+2 on IIM subgroups were similar, with ORs ranging between 2.1 and 3.7. Low C4T GCN had the greatest impact on IBM with OR=3.70 (2.72–5.04). Low C4A GCN had the largest impact on PM with OR=3.16 (2.65–3.75). Low C4L GCN had largest effects on PM and IBM, with ORs of 2.80 and 2.88, respectively.
C4 GCN variations among patients with IIM with and without MSA or MAAWe compared the mean age at diagnosis, sex and C4 GCN variations between patients with IIM with and without various myositis-related autoantibodies (table 3). Patients with anti-Jo1, anti-PM/Scl and MAA in general had younger age of disease diagnosis between 43 and 49 years old. Patients with IIM who tested positive for MSA or MAA were more likely to be women (70%–75%). Patients with anti-Jo1 and anti-PM/Scl consistently had the lowest mean GCNs of C4T, C4A and C4L.
Table 3Intragroup comparisons of demographics and complement C4 genes in patients with IIM with and without MSA or MAA
Except for anti-Jo1, patients with MSA presented with similar C4 or C3 plasma protein concentrations than those without. In contrast, patients with MAA had significantly lower levels of C4 and C3 than those without MAA (C4: 275.1±100.0 vs 330.9±105.4 mg/L, p=2.1×10−9; C3: 1188.0±309.8 vs 1335.2±283.9 mg/L, p=1.2×10−8). With regards to specific autoantibodies, patients with anti-PM/Scl and anti-Ro each had significantly lower C4 and C3 protein levels than those without these autoantibodies (figure 4A,B). Patients with MAA (83.6±30.6 vs 98.0±31.6 mg/L, p=1.1×10−7), anti-PM/Scl (86.1±26.7 vs 95.2±31.1 mg/L, p=0.03) and anti-Ro (85.0±30.1 vs 95.4±30.8 mg/L, p=0.014) had significantly lower C4 protein yield per gene (C4P/G) than those without these autoantibodies. No significant differences were observed on plasma protein levels of C4 and C4P/G between women and men (figure 4D,E).
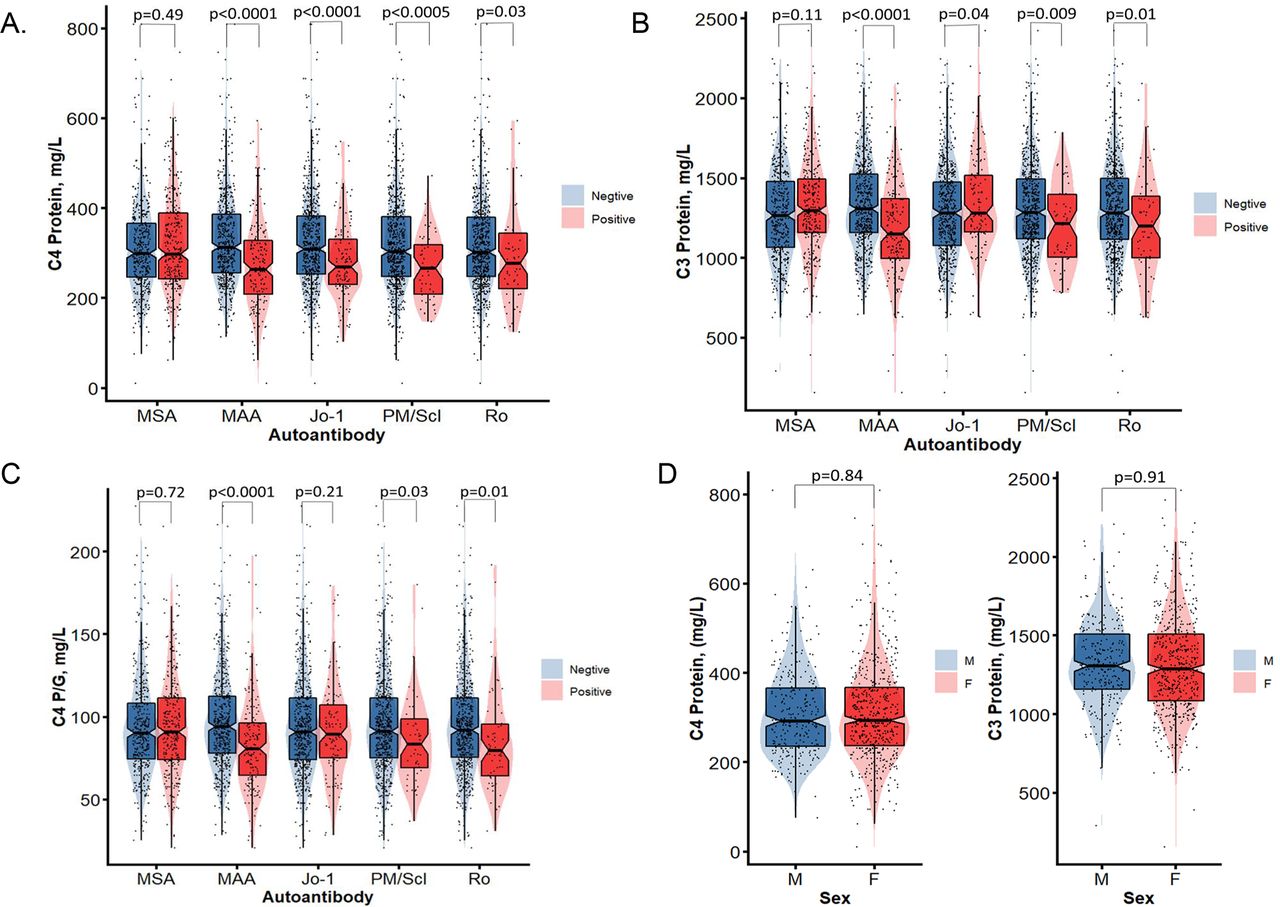 Figure 4
Figure 4 Comparisons of plasma protein levels for complement C4, C3 and C4 protein yield per C4 gene copies among patients with IIM with (+) and without (–) myositis-related autoantibodies including MSA or MAA in general, and anti-Jo1, anti-PM/Scl and anti-Ro (A, B and C). (D and E) Comparisons of C4 protein levels and C4/G between male and female patients with IIM. Violin plots are shown with median, 25th and 75th percentage range marked as boxes; red colour shades represent positive and blue colour shades represent negative with the specific autoantibodies of women and men, respectively, with p-values shown above. IIM, idiopathic inflammatory myopathies; MAA, myositis-associated autoantibodies; MSA, myositis-specific autoantibodies.
Logistic regression analyses of HLA-DRB1*03 and C4A deficiency in genetic risk of IIM and IIM-related autoantibodiesAmong healthy control subjects, 26.1% were HLA-DRB1*03 positive, compared with 56.1% in patients with IIM, which translated into an OR of 3.68 (2.94–4.60, p=2.6×10−32) in IIM (table 4). The distribution of HLA-DRB1*03 was uneven among subgroups of IIM, which varied from 75.4% in patients with IBM with an OR of 8.71 (5.48–13.8) to 59.5% in patients with PM with an OR of 4.16 (3.15–5.48), 47.6% in patients with DM with an OR of 2.57 (1.90–3.49) and 45.5% in patients with JDM with an OR of 2.36 (1.56–3.79).
Table 4Logistic regression models for genetic predictors in IIM, subgroups and autoantibodies
We performed logistic regression to investigate the relative roles of C4A deficiency and HLA-DRB1*03 as independent risk factors for IIM and subgroups. The results are shown in table 4. It was found that (1) C4A deficiency and C4 gene size variation were independent risk predictors of JDM and DM and (2) HLA-DRB1*03 and C4A deficiency and GCN of C4T were independent risk factors for PM and IBM. Moreover, HLA-DRB1*03 and C4A deficiency interacted to increase the risk of PM. We also performed intragroup logistic regression analyses to identify independent predictors of IIM-related autoantibodies. Complement C4 or C3 protein or C4P/G, HLA-DRB1*03 and/or HLA-DRB1*15, C4A deficiency or C4A GCN range of variations were risk factors for various myositis-related autoantibodies except for MSA in general. For patients with MSA, genetic factors such as HLA-DRB1*03, GCNs of C4B and C4L were independent predictors.
DiscussionHere we investigated complement C4 genetic diversity in patients with IIM of European descent and matched healthy controls. Our data consistently showed that low copy numbers of C4T and C4L, and C4A deficiency are highly significant risk factors for IIM and its major subgroups, with medium to large effect sizes45 or ORs between 1.7 and 3.7. Compared with healthy controls, patients with IIM had 0.28 to 0.58 fewer mean gene copies of C4T, C4A or C4L. The C4T=2 group yielded similar risks as the C4T=2+3 group, and the C4A=0 group had similar risk as the C4A=0+1 group. The similar magnitudes of ORs suggested that there were ‘dominant’ effects for low GCN of total C4 (ie, C4T=2 and C4T=2+3) and C4A deficiency (C4A=0 and C4A=0+1) on the risk of IIM, which is analogous to when homozygous and heterozygous mutants exhibit the same phenotype in Mendelian genetics. Such phenomena are in stark contrast to those observed in the genetics of human systemic lupus erythematosus (SLE), in which low GCNs of C4T or homozygous C4A deficiency (C4T=2, OR=6.51; C4A=0, OR=5.27) exerted substantially greater risks than those with C4T=3 (OR=1.32) or heterozygous C4A deficiency (C4A=1, OR=1.61).23 24 46 Parallel analyses of C4 structural variants between cases and controls recruited from each geographic location yielded similar results as presented for the entire IIM cohort, which are analogous to replication studies (online supplemental table S1).
Complement-mediated destruction leading to vasculopathy in dermatomyositis has been well-established,1 6 47 48 and we and others have demonstrated C4A genetic deficiency or low GCN of C4T in JDM.33 48 Demonstration of low C4T or C4L GCNs and C4A deficiency as genetic risk factors for DM, PM and IBM are novel findings of this work. These findings are relevant, as PM and IBM have been presumed to be disorders of cell-mediated immunity caused by target tissue cytotoxicity or destruction.1 The prevalence of low GCNs of C4T and C4L, C4A deficiency, and the presence of myositis-related autoantibodies in these diseases suggests that additional humoral immune effectors play a role in the pathophysiology of PM and IBM. IBM is unique as it has low GCNs in C4T, C4A and C4B. In a study of anti-Ro/anti-La patients with autoimmune diseases including myositis, Lundtoft and colleagues observed low GCNs of C4A in Scandinavian patients.49
It is worthy pointing out that the effects of GCN variation for C4S and C4B were opposite to those of C4L or C4A in JDM, DM and PM, which suggests different functions of C4S and C4B compared with C4L and C4A. Indeed, short C4 genes associate with higher C4 protein production50 51 and activated C4B protein generates faster activation of complement pathways52; long C4 genes associate with attenuated C4 protein production but possibly engage in antisense defence against viral infections.26 27 50 Moreover, activated C4A has greater efficiency to bind to immune complexes for clearance and protection against autoimmunity.26 27 50–53 We postulate that activated C4A and C4B proteins interfere and balance each other’s effects physiologically to achieve optimum defence against infections and autoimmunity and mitigate collateral damages due to complement-mediated injuries of self-tissue. C4A deficiency, which is also indicated by low copy numbers of C4T or C4L, and higher proportion of C4B among total C4 (C4B/C4T), would disturb such dynamic equilibrium and skew the immune response towards inflammation and autoimmunity with generation of autoantibodies.25 32
While IIM typically does not feature dramatic longitudinal fluctuations of plasma C3 and C4 protein levels with disease activity as is the case in SLE,54 intragroup analyses revealed that patients with IIM with anti-Jo-1, MAA in general, anti-PM/Scl or anti-Ro had significantly lower mean complement protein levels than those without these autoantibodies. Immune complexes formed by autoantibodies and self-antigens in IIM could activate and consume complement, leading to ‘depressed’ C4 and C3 plasma protein levels that were seen here and by others.55 Lower GCNs of C4T, C4L or C4A in IIM would be among the causes for lower C4 protein levels. Data on C4P/G yield, elevated levels of activation products in the plasma such as C4a, C3a and C5a, or cell-bound complement inactivation products such as erythrocyte-C4d and erythrocyte-C3d would help distinguish whether lower protein levels are due to genetic insufficiency or protein turnover.33 56 It is of interest to note that except for Jo-1, most MSA were not associated with lower complement levels in circulation, although MAA did. Moreover, patients with JDM have MSA such as anti-TIF1γ and anti-NXP24 57 and their relationship with complement activation is yet to be investigated.
In a study of complement in schizophrenia, SLE and Sjogren’s syndrome, it was suggested that C4 exhibited a sex-biased expression differences including in cerebrospinal fluid.58 We did not detect differential expression of C4 protein in EDTA-plasma between men and women among patients with IIM in this work or in previous studies.23 42 50 51 54 We did not detect differences in C4 GCN variations between female and male patients for DM, PM and JDM (online supplemental table S2). However, IBM is a male-dominant disease and we observed slightly higher frequencies of low GCNs for C4L, lower proportions of C4A or C4L among C4T in men compared with women.
The relative roles of HLA class II variants including DRB1*03, DQA1*05, DQB1*02 and C4A deficiency on genetic predisposition to autoimmune diseases such as IIM are an unsolved enigma.17 47 59 Multivariate logistic regression analyses revealed that C4A deficiency was an independent risk factor for DM and JDM and that HLA-DRB1*03 was a prominent risk factor for IBM, while C4A deficiency and HLA-DRB1*03 contributed independently and interactively to an increased risk of PM. Further analyses of DRB1, DQA1, DQB1 variants and GCNs of C4 revealed the presence of both risk and protective factors in each gene on the predisposition of IIM subgroups and autoantibodies (online supplemental figure S3 and online supplemental table S4).
In summary, our results demonstrated that low GCNs for C4T, C4A and C4L played significant roles in increasing the risk of IIM. The relationship between C4A deficiency and HLA-DRB1*03, which are closely linked, is complex and intriguing. It will be important going forward to carefully interrogate the mechanisms by which HLA-DRB1*03 and C4A deficiency contribute to autoimmunity and IIM. Finally, intragroup analyses showed that patients with IIM with certain autoantibodies presented with lower protein levels of complement C3 and C4. This effect was more notable for MAA than for MSA, which is worthy of investigations. Our findings have broad implications in the assessment and treatment of IIM and other autoimmune diseases.
Data availability statementData are available on reasonable request.
Ethics statementsPatient consent for publicationEthics approvalThis study involves human participants and was approved by the Institutional Review Board (IRB) at the Nationwide Children’s Hospital, and the Committee of the Myositis Genetics Consortium (MYOGEN). Our IRB approval number is IRB14-00544: MHC and CNVs in Disease Associations. Participants gave informed consent to participate in the study before taking part.
AcknowledgmentsWe are indebted to volunteers and patients with myositis who contributed blood samples for this study, and to Bi Zhou and Alex Sepeda for technical assistance. We thank Drs Daniella Schwartz and Pravitt Gourh for valuable comments on the manuscript.
留言 (0)