Reagents and antibodies
1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 1,2-dilauroyl-sn-glycero-3-phosphocholine (DLPC) were purchased from Avanti Polar Lipids (Alabaster, AL, USA). Polyethylene glycol (PEG, MW 8,000), polyethyleneimine (PEI branched, MW 25,000), Complete Freund’s adjuvant (CFA), Incomplete Freund’s adjuvant (IFA), and methylthiazolyldiphenyl-tetrazolium bromide (MTT) were purchased from Sigma‒Aldrich (St. Louis, MO, USA). Anti-mouse CTLA4 monoclonal antibody (mAb) and anti-mouse CD28 mAb were purchased from BioLegend (San Diego, CA, USA). Anti-human CD3 mAb (OKT3) and anti-mouse CD3 mAb (2C11) were kindly provided by Dr. Steve R. Roffler (Institute of BioMedical Sciences, Academia Sinica, Taipei City, Taiwan, ROC).
Others
Peptide-HLA-A2 monomer and one identify epitope of HPV type 16 E7 protein (YMLDLQPETT) were kindly provided by Dr. Shih-Jen Liu (NHRI) [24]. Additionally, TC1/AAD cells were gifts from Dr. Shih-Jen Liu (NHRI).
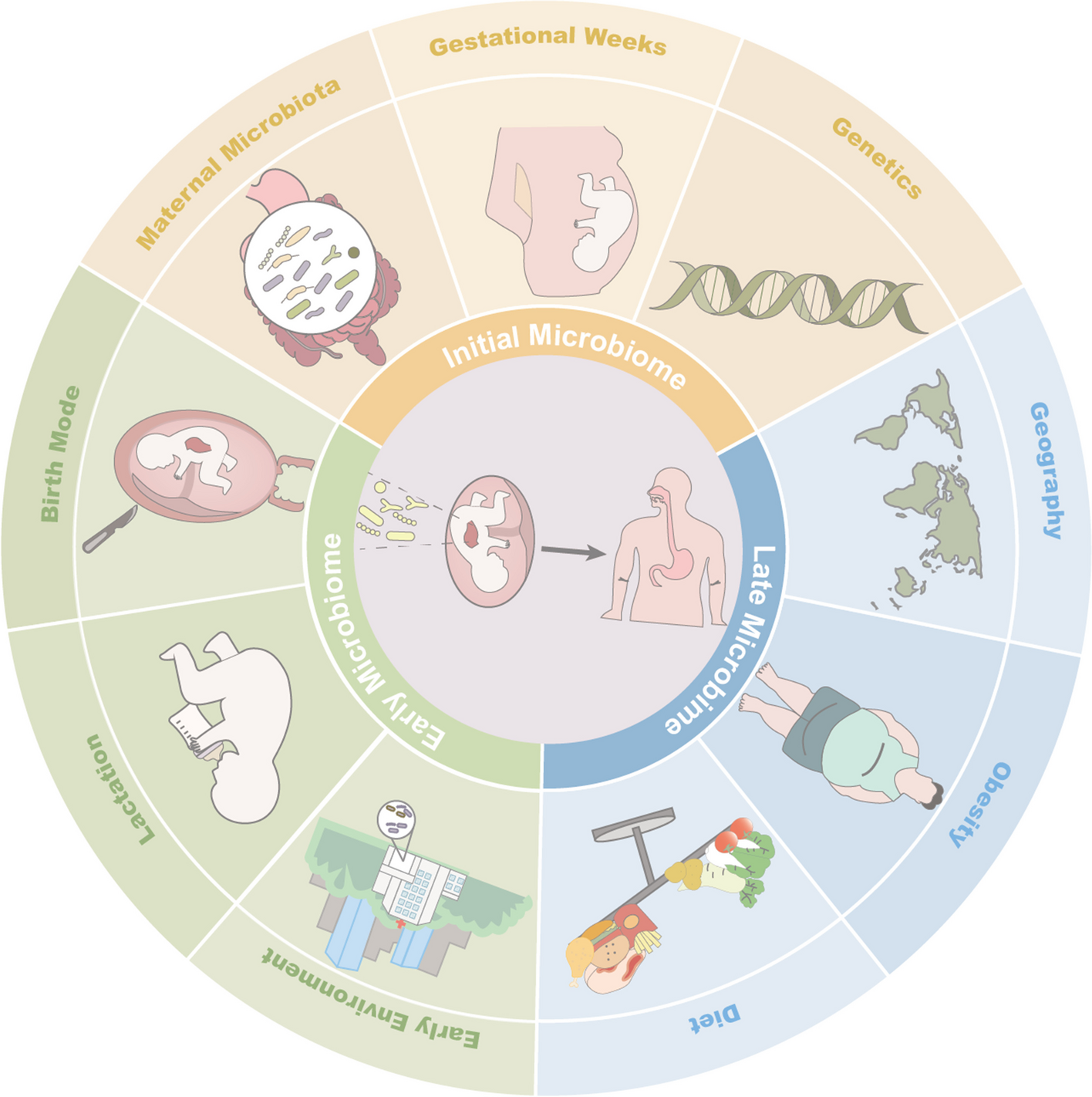
Analysis of LPPC size and surface charge
LPPCs were prepared according to the procedures outlined in previous studies [14, 20]. The particle size and the surface charge of LPPC were analyzed by a Zetasizer (Zetasizer Nano ZS90, Malvern, UK). Briefly, ten micrograms of LPPC were complexed with or without immune molecules, such as 0.15 µg anti-CD28 mAb, 0.15 µg anti-CTLA4 mAb, 20 µg dendritic cells membrane proteins or 0.6 µg HLA-A2 protein. After adsorption, the addition of PEG into immuno-LPPC complexes neutralized the amine group of PEI from the LPPC components. The different combinations of immuno-LPPC complexes were listed in Table 1. LPPC complexes were applied onto a glow-discharged TEM grid (300-mesh Cu grid) coated with a holey carbon film. The excess liquid was blotted, and the specimens were vitrified by rapid plunging into liquid ethane precooled with liquid nitrogen. Cryo-TEM was performed on a CEM2 instrument (Tecnai G2 F20 TWIN) at Academia Sinica, Taiwan.
Table 1 The particle sizes and surface charges of LPPC in combination with immunofunctional molecules. LPPC adsorbed different mAbs and proteins in the following experiments. The addition of PEG for charge neutralization was represented as w/, otherwise, it was shown as w/o. A Zetasizer was used to analyze the characteristics, particle size, and zeta-potential of LPPC with different molecules. All values were the mean ± SD of three independent experiments (N = 6)The capacity and activity of LPPC with mAbs
Ten micrograms of LPPC were incubated with different amounts of OKT3 or 2C11 for 20 min and centrifuged at 5,900 × g for 5 min. The pellets were resuspended, and the quantity of OKT3 or 2C11 bound to LPPC was determined by using a Coomassie Plus Bradford Assay Kit (Sigma‒Aldrich) according to the standard procedures provided by the manufacturer. The supernatants were analyzed as described above.
Peripheral blood mononuclear cells (PBMCs) were seeded in a 96-well plate at 1 × 105 per well in 100 µl RPMI medium containing 10% FBS and 1% PSA. LPPCs (1µγ) with different amounts of mAbs were added respectively to each well of a 96-well plate. After incubation for three days, the cell proliferation rates were determined by the MTT assay (Sigma‒Aldrich).
Similarly, in the naive group, splenocytes (2.5 × 105 cells, each well of a 96-well plate) from naive mice were incubated with 1 µg LPPC with different amounts of mAbs for three days. In the activated groups, splenocytes from naive mice were activated by anti-CD3 (60 ng/100 µl) and anti-CD28 mAbs (60 ng/100 µl) for three days. After stimulation, the splenocytes were reseeded into a 96-well plate. The cell proliferation rate was monitored by MTT assay.
Real-time PCR
Naive or activated splenocytes prepared as described above were incubated with LPPC, LPPC/CD3, LPPC/CD3/CD28, or LPPC/CD3/CTLA4 for 12 h, and approximately 1 × 106 cells were centrifuged at 400 × g for 5 min. Then, total RNA was extracted from the cell pellets using TRIzol reagent (Invitrogen, Gaithersburg, MD, USA) following the manufacturer's instructions.
All real-time PCR reactions were performed using an ABI Prism 7000 Sequence Detection System (Perkin-Elmer Applied Biosystems, Foster City, CA, USA). The 2(-ΔΔ)(CT) method was used to calculate relative changes in gene expression as determined by real-time quantitative PCR experiments. In the present study, the data are presented as the fold change in target gene (IL-2) expression in splenocytes normalized to the internal control gene (β-actin).
Caspase 3 activity
Naive or activated splenocytes were incubated respectively with different treatments for 4 h, and then the caspase 3 activity of the cells was determined by the PE-active caspase 3 apoptosis kit (Becton Dickinson, Mountain View, CA, USA) and analyzed by flow cytometry.
Cell cycle
Naive or activated splenocytes were incubated with different treatments for 48 h, as in a previous study [15], the 1 × 106 cell pellets were washed with PBS and fixed in 3 ml of 70% ethanol at 4 °C for 30 min. After centrifugation at 400 × g, the fixed cells were resuspended in 1 ml of DNA staining buffer (containing 5% Triton-X 100, 0.1 mg/ml RNase A and 4 μg/ml propidium iodide) and incubated for 30 min at room temperature. Ten thousand cells were analyzed for DNA content using a Becton–Dickinson FACScan (Becton Dickinson, Mountain View, CA), and the cell cycle distribution was determined using ModFit software (Becton Dickinson).
The activities of the LPPC/HLA/Ab complex in vivo and ex vivo
The transgenic mice were vaccinated by IV injection with 6 µg peptides-loaded HLA-A2 and 1.5 µg anti-CD28 mAb on 100 µg LPPC; 6 µg peptides-loaded HLA-A2 on 100 µg LPPC; 100 µg LPPC; 6 µg peptides-loaded HLA-A2; 6 µg peptides-loaded HLA-A2 and 1.5 µg anti-CD28 mAb; or PBS. After two weeks, the mice were individually boosted with the same treatments. Then, one week later, the activity of the immuno-LPPCs was tested. The splenocytes isolated from the mice were cocultured with YML peptides (2 μg/ml) in a 96-well plate at 2.5 × 105 cells per well. The MTT assay was used to estimate the proliferation of cells at 72 h, and ELISA measured the cytokine concentrations of the supernatants.
The specific activities of the LPPC/HLA/Ab complexes (0.6 µg peptides-loaded HLA-A2, and 0.15 µg anti-CD28 or anti-CTLA4 mAb on LPPC) react to splenocytes, the mice prior immunized with YML peptides (30 µg) and Freund's adjuvant, were determined based on cell proliferation and cytokine expression.
Cytotoxicity assay (LPPC/HLA/Ab complex)
Mice were immunized and boosted with immuno-LPPCs as described above. On the 21st day after immunization, splenocytes from treated mice were used as effector cells. In a 96-well plate, TC1/AAD cells as target cells (1 × 104 cells/well) were seeded overnight, and splenocytes were added into the separated wells at E:T ratios of 100:1, 50:1, 25:1, and 12.5:1. After 72 h, the supernatants were removed, and the cells were washed with PBS six times. The MTT assay determined the lysis percentage of target cells, which was calculated by [survival rate of nontreated group (100%)—survival rate of the individual group].
For analysis of immunomodulation efficacy of LPPC/HLA/Ab complexes on splenocyte cytotoxicity, the mice were first immunized and boosted with YML-peptides for three weeks. Then, the mice were treated with LPPC/HLA/Ab complexes once weekly for two weeks. In the 5th week, the MTT assay evaluated the cytotoxicity of the splenocytes against TC1/AAD cells.
Preparation of lipoplexes with DC membrane proteins and Abs
A previously described method for the isolation and culture of DCs was used [25]. DC membrane proteins (MP) were extracted using a Mem-PER Eukaryotic Membrane Protein Extraction Reagent Kit (Thermo Fisher Scientific Inc., Waltham, MA, USA). For DC MP adsorbed with LPPC, LPPCs (100 µg) were incubated respectively with different amounts of membrane proteins in one tube for 30 min and then centrifuged. The supernatants were removed, and the pellet was resuspended in 100 µl of RPMI solution (Invitrogen).
To form LPPC/MP/Ab complexes, 100 µg of LPPC, different amounts of MP (50, 100, 200 µg), and 1.5 µg anti-CD28 or anti-CTLA4 Abs were co-incubated in one tube for 30 min and centrifuged to remove the supernatants. The pellet was resuspended in 100 µl of RPMI solution for further investigation.
Immunization with LPPC/MP complex or immunomodulation with LPPC/MP/Ab complex in vivo
Six- to eight-week-old female BALB/c and C57BL/6 mice were purchased from NLAC. The mice were immunized by IV injection with the different treatments, such as 200 µg MP on 100 µg LPPC; 200 µg MP and 1.5 µg anti-CD28 mAb on 100 µg LPPC; 200 µg MP and 1.5 µg anti-CTLA4 mAb on 100 µg LPPC. After two weeks, mice were boosted with the same complex by IV injection. Moreover, under immunomodulation treatments, mice that were immunized and boosted with LPPC/MP complexes were by IV injection of different LPPC/MP/Ab complexes weekly.
ELISPOT (LPPC/MP/Ab complex)
Mice were immunized and boosted with immuno-LPPC complexes as described above. The investigation of the activity of LPPC/MP complexes was performed by collecting splenocytes from the treated mice 21 days after immunization, and the number of cells expressing IL-4 or IFN-γ was monitored using a U-CyTech BV mouse ELISpot kit (IFN-γ or IL-4; U-CyTech, Yalelaan, The Netherlands) following the manufacturer’s standard protocol. The spots in the wells were visualized and counted using an inverted microscope (Olympus, Shinjuku, Tokyo, Japan).
Cell proliferation and cytokine ELISA (LPPC/MP/Ab complex)
The mice were immunized and boosted as described above. Splenocytes from treated mice were pulsed with HpHsp60 antigen (1 µg/ml) or EGFP antigens (1 µg/ml) for 72 h, an MTT assay (Sigma‒Aldrich) was estimated cell proliferation, and then the supernatants were analyzed for cytokines (TGF-β, IL-4, and IFN-γ) using ELISA (R&D, Minneapolis, MN, USA).
Cytotoxicity (LPPC/MP/Ab complex)
As described in our previous study [26], BALB/3T3 cells were transfected with pCJ-H. pylori heat shock protein 60 (HpHsp60) or pCJ-HpUreaseB (provided by Dr. Wu, National Taiwan Ocean University, Keelung City, Taiwan, ROC), which expressed HpHsp60 or Urease B, respectively. The transfected BALB/3T3 cells were plated (1 × 104 cells/well in a 96-well plate) and then incubated overnight. Splenocytes obtained from the immunized mice were added to the wells as effector cells at E:T ratios of 100:1, 50:1, 25:1, and 12.5:1. After incubation for 72 h, the cell viabilities were determined by an MTT assay.
B16F10-pLEGFP or B16F10 cells were seeded at 2 × 103 cells/well in a 96-well plate and incubated overnight. Splenocytes were added to the wells as effector cells at an E:T ratio of 100:1. After incubation for 72 h, the cell viabilities were determined by the MTT assay.
H&E staining
As described previously [21], the organs or tissues of the mice were formalin-fixed, paraffin-embedded and subsequently stained by using histochemical techniques. The tissue slides were observed under a light microscope.
RNA sequence analysis of mice splenocytes
The mice were immunized and boosted by LPPC/MP complexes with different antibodies, and the RNA collection of procedure was shown in Fig. 9A. Splenocyte RNA was extracted using a PureLinkTM RNA Mini kit (Thermo Fisher Scientific Inc., Waltham, MA). Then, NGS was performed with a NovaSeq 6000 system (Illumina, CA). The raw counts were estimated and normalized as fragments per kilobase per million (FPKM). The gene expression was further selected by t-test and ANOVA analysis. All supporting NGS data were available to researchers in the Gene Expression Omnibus (GEO) database, and the accession number was GSE217167.
Statistical analysis
Data were analyzed using the SAS statistical software package (SAS Institute, Inc., Cary, NC, USA). Two independent trials were compared by a t-test, and multiple variables were compared by ANOVA. P < 0.05 indicated statistically significant differences. All results are expressed as the mean ± SD.




留言 (0)