
記住我


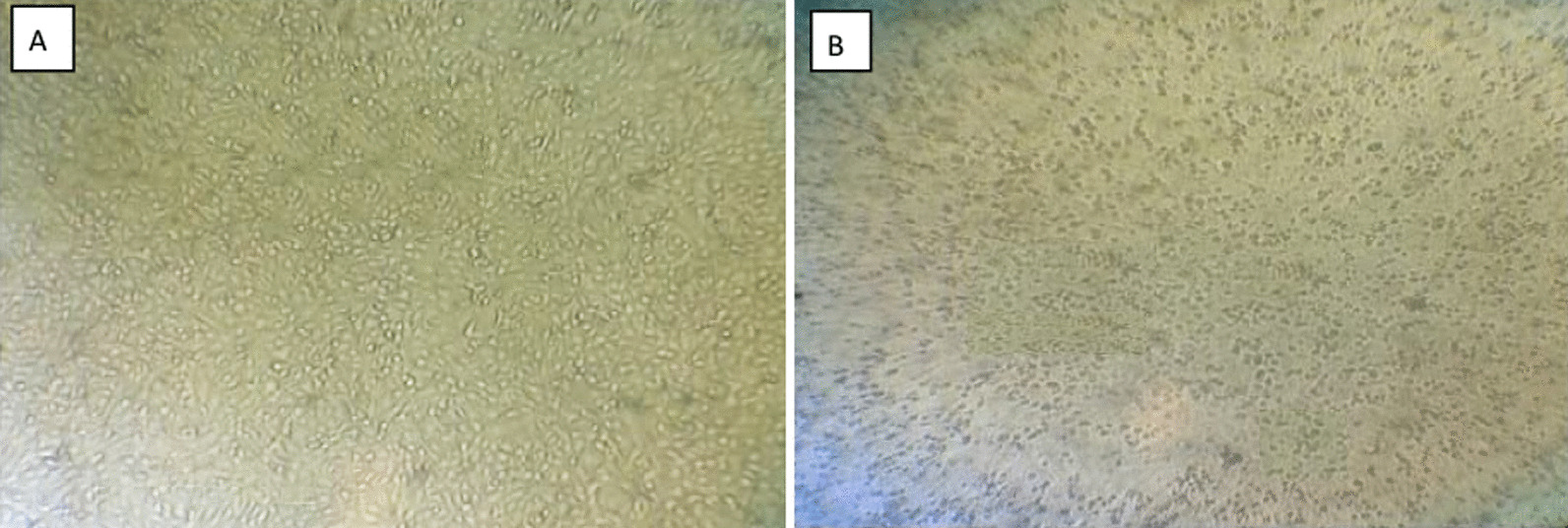
The total RNA was extracted from the small intestine and its contents in order to determine the diarrhea antigen collected in the pig farm in Heilongjiang province in China, and the RT-PCR result was positive for the PEDV (Fig. 1 A and Additional file 1: Supplementary Table S1). The sample was homogenized to produce an adsorbent, infected with Vero E6 cells, and RT-qPCR was applied to the first venom, with domesticated CV777 used as a positive control (Fig. 1B). CPEs were found upon virus blind passage of the twelfth generation, with the concomitant formation of syncytia as well as cell shedding (Fig. 1C). After 24 h of isolation in Vero E6 cells, IFA showed specific fluorescence with monoclonal antibody against the PEDV N protein, and there was no PEDV in the negative control (Fig. 1D). As a result of the quantitative analysis, CV777 was used as a positive control, and the isolate was adapted. Based on the above results, we named CH/HLJJS/2022. The TCID50 was measured by the Reed-Muench method, and a one-step growth curve was drawn, showing that this strain was significantly more virulent than the classical strains at the virulence level (Fig. 2A). It was confirmed by microscopic observation of infection 60 h (Fig. 2B). To better explain this phenomenon, we constructed ORF3 recombinant plasmids targeting conserved sequences of PEDV and plotted a standard curve (Fig. 2C). Viral vector measurements were performed for 60 h, 72 and 84 h of Vero E6 cells, and the results were compatible with the growth curves (Fig. 2D). CH/HLJJS/2022 was more virulent than the classical strains before 72 h, and the virus particles were slowly inactivated and likely degraded in the incubator at 37 °C, possibly due to host death after 72 h.
Fig. 1
A Assays for PCV2, PDCoV, TGEV, PRRSV, PBoV, PRV, PKV, PEDV, BVDV were performed sequentially. B The 96 h viral culture fluid, which had been domesticated CV777 as a positive control, was examined. C Microscopic observation over 12 generations of blind passage. D Immunofluorescence results were obtained at 24 h of infection (n = 3)
Fig. 2
A Plot the one-step growth curve of the virus. B Infection by the isolated viruses passaged up to passage 12 was observed microscopically for 60 h. C ORF3 recombinant plasmids were prepared to generate a standard curve. D Viral load assays were performed at 60 h, 72 and 84 h of CH/HLJJS/2022 infection (n = 3, P > 0.005)
Complete genome sequence of CH/HLJJS/2022The complete genome sequence of CH/HLJJS/2022 was deduced using Illumina platform and submitted to GenBank with the login number ON968723.1 (Additional file 1: Fig. S1). A total of 28,097 nucleotides were detected for this strain, including ORF1a (nt 300–12,653), ORF1b (nt 12,683–20,644), S (nt 20,641–24,801), ORF3 (nt 24,801–25,475), E (nt 25,456–25,686), M (nt 25,694–26,374), and N (nt 26,386–27,711). The full gene phylogenetic analysis showed that CH/HLJJS/2022 belongs to the GIIa subtype (Fig. 3 A and Additional file 1: Table S2), which is consistent with the developmental analysis of the S gene (Fig. 3B). It has also been maintained at some distance from classical strains in evolutionary analyses of ORF3, E, N, and M proteins, although the N protein is relatively conserved and there are no differences in small protein M (Additional file 1: Fig. S2). Genome wide homology analysis indicated that CH/HLJJS/2022 shared 96.1−98.9% identity with other strains and was most similar to T10-HB2018, while CV777 was shown to share 97.9% nucleotide homology (Additional file 1: Figs. S2 and S3). The homology normalized heat map shows significant differences when compared to the genomes of other strains, and this result was repeated for S and other proteins (Fig. 4 and Additional file 1: Fig. S4).
Fig. 3
The whole genome and S protein sequences of the CH/HLJJS/2022 strain were analyzed for genetic evolution. A The CH/HLJJS/2022 and 49 PEDV strains were subjected to evolutionary analysis and divided into two subtypes, GI and GII, with GIa, GIb and GII further subdivided into GIIa, GIIb, GIIc. B S protein is the same as above
Fig. 4
Analysis of the entire genome and homology of the S protein sequence of strain CH/HLJJS/2022. A The whole genomes of 50 viruses were analyzed for homology, and the homology results were subjected to a normalized Heatmap. B The S protein was processed as above. P ≤ 0.05 were considered statistically significant in the Heatmap
Amino acid mutations of CH/HLJJS/2022To determine the specificity of CH/HLJJS/2022, amino acid alignments and analyses were performed using DNAMAN on two randomly selected GIa, GIb, GIIb, and GIIc strains and one additional GIIa strains. The results showed that eight amino acid mutations, N-D (aa 139), P-L (aa 229), I-T (aa 287), T-N (aa 394), F-L (aa 539), L-M (aa 998), P-S (aa 1264) and E-K (aa 1299), were generated located on the S protein (Fig. 5 A and Additional file 1: Fig. S5). Three, one and one amino acid mutations, L-I (aa 18), Q-H (aa 250), N-S (aa 441), V-I (aa 129), and N-D (aa 46) resulted from being located in the N, M and E proteins, respectively (Fig. 5B–E and Additional file 1: Fig. S6).
Fig. 5
Two strains of each subtype were selected for comparison. A S protein to contrast. B ORF3 protein to contrast. C N protein to contrast. D M protein to contrast. E E protein to contrast. The red box represents the characteristics of each subtype, and the blue box represents the unique evolutionary characteristics of CH/HLJJS/2022
The results also showed that there was a certain amount of amino acid specific conservation, including S, ORF3, E, and M proteins, among the grouped strains, and these phenomena were the basis for the identification of PEDV grouped strains. Examples are GI/GII group LRS/LQS (aa 1305–1307) in the S protein, GIIa b/c group SANT/QSTI (aa 27–30), GIa/GIb and GII group SVN/SAN (aa 20–22) in the ORF3 protein, GI/GII group MRI/MQI (aa 64–66) in the E protein, GI/GII group SVF/SAF (aa 38–40) in the M protein (Fig. 5). The segregation of CH/HLJJS/2022 implies that novel mutations have arisen in PEDV to include proteins traditionally thought to be highly conserved. While reducing primer accuracy for previous typing assays.
Mutations of 3-aa altered S protein spatial structureRepresentative strains from each subtype were selected for protein spatial structure modeling, and structural alterations in the S and M proteins were identified through five sets of subtypes and CH/HLJJS/2022 alignments (Additional file 1: Fig. S7 and Fig. 6). No significant changes were found for ORF3, E and M proteins. To further identify alterations brought about by unique amino acid mutations, we narrowed the simulations to identify specific changes at the mutation site. The results showed that three mutations in the S protein altered the tertiary structure, N-D (aa 139), F-L (aa 539) and P-S (aa 1264) (Fig. 7A, C and E). At the same time, we found that highly conserved amino acids within the subtypes produced divergent structures, consistent with our typing landmark structure derived from our sequence alignment above (Fig. 7B, D and F). To ensure the accuracy of the conclusions, we established the tertiary structure by SWISS-MODEL, which was verified using Phyre2 and FirstGlance in JMOL (Data not shown).
Fig. 6
Protein modeling contrasts between CH/HLJJS/2022 and other subtype strains (AJ1102, MEX/124/2014, OH851, CV777 and PPC14). A The S protein contrasts with the finding of markedly differential changes in the first 500 amino acids. B M protein model. C E protein model alignment results. D ORF3 protein model. (N) N protein model
Fig. 7
Detailed alignment of the S protein model to the reference strain A Structural changes caused by mutation of specific amino acid between CH/HLJJS/2022 strain of S protein and other strains. B Structural differences due to grouping landmarks between the various subtypes of S (aa 1–400) protein. C–F A similar situation
The 4-aa alters the S antigen epitopeOf the eight unique mutations in the S protein found, the S1° and COE neutralizing epitopes were involved. The CH/HLJJS/2022 specific antigenic epitope was found to be distinguished from other strains using DNASTAR, after alignment of the S protein sequence specificity and structural specificity. N-D (aa 139), P-L (aa 229), I-T (aa 287) and L-M (aa 998) showed epitope differences from currently identified PEDV (Fig. 8A).
Fig. 8
Protein function prediction. A Primary epitope comparative analyses of five subtype representative strains and CH/HLJJS/2022 strain revealed 4-aa mutations out finding specific changes. B S protein transmembrane function prediction. C Prediction of M protein transmembrane function. D N protein phosphorylation prediction
Although the S protein mutations altered the antigenic epitope compared to CV777, there were no actual changes in transmembrane function (Fig. 8B). The same is true for the transmembrane function prediction of the M protein (Fig. 8C). N-protein S-terminus of CH/HLJJS/2022 allowed enrichment of phosphorylation functions compared to the N-terminus of the other isoforms, but was not revealed due to a prediction score of 0.48 (P < 0.5, Fig. 8D).
留言 (0)