Animals
All C57BL/6 male mice (6–8 weeks) were purchased from the Animal Experimental Center of the Fourth Military Medical University, and all mice were raised in a standard environment as described previously [15]. The whole animal experiment scheme has been approved by the Animal Ethics Committee of the Fourth Military Medical University (approval no. IACUC-20210358).
Extraction and culture of BMDMs
C57BL/6 mice were killed and sterilized in 70% ethanol for 15 min. The intact femur and tibia of the hindlimb of mice were removed with sterile instruments. The bone marrow cavity was washed repeatedly with precooled phosphate-buffered saline (PBS), and the completely mixed suspension was filtered and collected in a 15 mL centrifuge tube. Red blood cell lysate was added in the ratio of 1:3 to lyse for 10 min. After centrifugation (300g × 5 min), the supernatant was discarded, and the cells were gently resuspended and cultured in a modified Dulbecco medium containing 10% fetal bovine serum and 10 ng/mL macrophage colony-stimulating factor (MCSF). All cells were cultured in 37 °C incubator containing 5% CO2 for 7 days until maturation. M1 polarization of BMDMs was induced by LPS (100 ng/mL, Sigma-Aldrich, USA) + INF-γ (20 ng/mL, PeproTech, USA).
Immunofluorescence
The cells were fixed at room temperature with 4% paraformaldehyde for 20 min. The frozen sections of spinal cord tissue or cells were washed with PBS three times, and then incubated with 0.3% Triton X-100 for 30 min. Bovine serum albumin (BSA) was used to block for 30 min and incubated overnight with primary antibodies at 4 °C. The primary antibodies used include anti-F4/80 (cat. no. ab6640, Abcam,1:300), anti-iNOS (cat. no. 13120, Cell Signaling Technology, 1:300), anti-MAP2 (cat. no. 8707, Cell Signaling Technology, 1:300), anti-β-III-tubulin (cat. no. ab78078, Abcam, 1:300), and anti-NeuN (cat. no. ab177487, Abcam,1:300). The next day, the second antibodies was incubated at room temperature for 1 h and the nucleus was stained with DAPI. Finally, the fluorescence image was obtained under a fluorescence microscope (BX51, Olympus).
Flow cytometry
The cells were collected after 48 h of treatment and resuspended with PBS. Under dark conditions, F4/80 antibody (APC-F4/80, 1:50, eBioscience, cat. no. 17-4801-82) was added to M0 macrophages, F4/80 and CD86 antibody (PE-CD86, 1:200, BioLegend, cat. no. 105014) were added to M1 macrophages, and then incubated for 30 min at 4 °C. Identification and detection of macrophages by flow cytometry (Beckman Coulter, CA, USA).
Transcriptome sequencing analysis
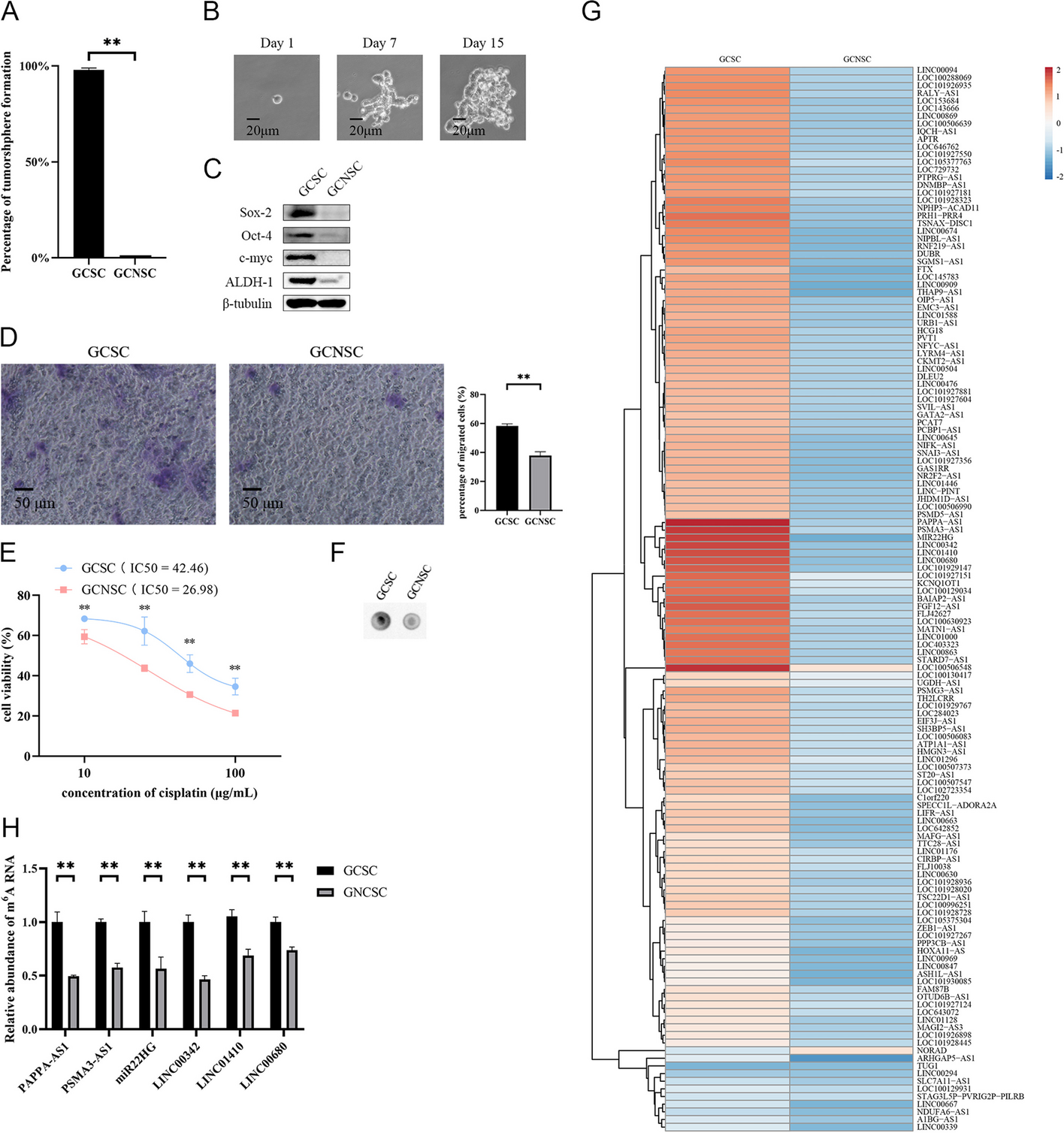
BMDMs were harvested after treatment, and total RNA was extracted with TRIzol reagent. Our sequencing was divided into three groups, including M0 group, M1 group, and M1 + PBM group (n = 3 per group). Genergy Biotechnology Co. Ltd (Shanghai, China) performed enrichment, fragmentation, reverse transcription, library construction, sequencing, and data analysis. Fastq-formatted raw data were processed and analyzed. The number of transcripts in each sample was calculated according to fragments per kilobase of transcript per million fragments mapped (FPKM). For each sample, FPKM values were calculated using Cuffnorm software, and log2 transformations were applied. The differential gene expression between different samples was calculated using DESeq2 software. The threshold of differentially expressed transcripts was determined to be P < 0.05 and multiple change ≥ 1. The KEGG database was used to analyze the signal pathway enrichment of differentially expressed transcripts. When P < 0.05 and at least two genes are involved, the pathway of significant enrichment can be determined. The STRING database (https://string-db.org/) is used to construct PPI networks for DEmRNAs, download interaction data, and analyze hub genes with Cytoscape software. miRNAs-target lncRNA TUG1 and miRNAs-target mRNAs were predicted by starBase database (https://starbase.sysu.edu.cn/). Comprehensive ceRNA score and expression value prediction results were used to screen ceRNA. The main data were uploaded to the NCBI database (login number PRJNA780778).
Cell transfection
The knockdown and overexpression adenovirus of TUG1 was synthesized by Hanbio Biotechnology (Shanghai, China), and the optimal multiplicity of infection (MOI) was selected for transfection. TLR3 siRNA, miRNA-1192 mimics and inhibitors were purchased from GenePharma Co. Ltd (Shanghai, China). Lipofectamine 2000 was used as transfection reagent. Poly(I:C) (Sigma-Aldrich, MO, USA) was used to stimulate the expression of TLR3. All reagents are transfected according to the manufacturer’s instructions.
Cytoplasmic and nuclear fractionation
According to the manufacturer’s instructions, Minute Cytoplasmic and Nuclear Extraction Kits (Invent Biotechnologies, Berkshire, Plymouth, USA) were used to separate the cytoplasm and nucleus of macrophages. The isolated extract was dissolved with TRIzol reagent to extract RNA, and the subcellular localization of TUG1 was detected by RT-PCR. GAPDH and U6 were used as cytoplasmic and nuclear controls, respectively.
SCI model
The SCI model is constructed as previously described [23]. Mice were anesthetized by intraperitoneal injection of 0.6% sodium pentobarbital, and their back hair was removed. The mice were fixed on a sterile operating table and disinfected with 75% medical alcohol. T9 was taken as the center to make a longitudinal incision. The skin and subcutaneous tissue was cut in turn, the spinous process and lamina of T8–T10 were exposed, and T9 lamina under microscope was removed to completely expose the spinal cord. The modified forceps was used to clamp spinal cord tissue (vertical direction) for 30 s to cause SCI, then hemostasis and suture. After SCI, manually squeeze the bladder to urinate every day, and observe the vital signs of mice. The control group only underwent laminectomy.
Photobiomodulation therapy
SCI mice were randomly divided into PBM treatment group and injury group. The mice were anesthetized by intraperitoneal injection of 0.6% sodium pentobarbital and placed in a dark cage (temperature 25 °C). An 808 nm laser device (MW-GX-808/1000 mW, near-infrared spectrum) made by Changchun Leishi Optoelectronic Technology Co., Ltd and its supporting medical diffusion optical system were used for PBM treatment in mice. The safety and irradiation parameters of optical fibers have been verified in piglets [14, 24]. The medical highly transparent silica coating on the surface of optical fiber ensures its flexibility and biocompatibility without affecting its optical properties. The optical fiber is cylindrical with a diameter of 600 μm. Use a calibrated optical sensor to confirm that the output power of the optical fiber is consistent with the set power. We irradiated the spinal cord injury area of mice for 50 min every day (50 mW/cm2). At the cellular level, the cells are placed on an ultraclean worktable and exposed to 808 nm low-level laser irradiation every 12 h. The specific parameters are described in Additional file 2.
Quantitative real-time PCR
After the cells were treated (the spinal cord tissue was removed and ground), the total RNA was extracted with TRIzol reagent according to the manufacturer’s instructions. cDNA was obtained using Evo M-MLV RT premix reagent (AG11706, Accurate Biotechnology, China). The reaction conditions are 37 °C, 15 min, 85 °C, 5 s, and 4 °C 10 min. SYBR Green is used for quantitative real-time PCR (qPCR). CFX (Invitrogen, Waltham, Ma, USA) and CFX connect real-time PCR system (BioRad, Hercules, CA, USA) were used for 15 s at 95 °C, followed by 40 cycles at 95 °C for 5 s and 34 s at 60 °C. Use the 2−ΔΔCT method to analyze the data. miRNA-1192 primers and the internal reference U6 were synthesized by General Biology Co., Ltd. All primers are listed in Additional file 3.
Western blotting analysis
The cells were washed with PBS and then lysed on ice with RIPA buffer containing phosphatase inhibitor (the spinal cord tissue in the injured area was fully ground after adding RIPA buffer containing phosphatase inhibitor). All proteins were harvested and transferred to a 1.5 mL centrifuge tube after 20 min of cleavage, and the precipitate was discarded after 20 min of centrifugation (4 °C × 12,000g). BCA protein analysis kit is used to detect protein concentration (Thermo Scientific, 23227). The total protein extract was separated by SDS-PAGE and transferred to nitrocellulose membrane (P-N66485, Pall, America). The nitrocellulose membrane was sealed at room temperature for 1 h in 5% skim milk and then incubated overnight with primary antibody at 4 °C. Primary antibodies: iNOS (cat. no. 13120, Cell Signaling Technology, 1:1000), TLR3 (cat. no. ab62566, Abcam, 1:1000), p-NF-κB (cat. no. 3033, Cell Signaling Technology, 1:1000), and β-actin (cat. no. 66009-1-IG, Proteintech, 1:3000). The next day, the secondary antibody was incubated at room temperature for 1 h, and the Amersham Imager 600 (General Electric) was used for imaging after adding ultrasensitive luminescent solution.
Neuronal culture and treatments
The dorsal root ganglion (DRG) was extracted from Sprague Dawley rat neonates (P1–P3) using the method we previously reported [25]. The DRG was completely cut, digested with trypsin digestion solution (0.125%) and type IV collagenase solution (0.1%) for 30 min, and then supplemented with 20% FBS DMEM–F12 to stop digestion. The cells were centrifuged at 1000 rpm for 5 min and then suspended in a medium with addition of B27 and 1% penicillin/streptomycin and cultured in a 12-well plate.
To explore the effect of BMDMs on DRG toxicity, we collected the culture supernatant of BMDMs, including M0 macrophage-conditioned medium (MCM), M1-MCM, shTUG1-MCM, shTUG1 + PBM-MCM, OE-TUG1-MCM, and OE-TUG1 + PBM-MCM. The conditioned medium was filtered with 0.22 mm membrane to remove the cell residue. Half of the medium of DRG was replaced by MCM. After neurons were cultured in mixed medium for 24 h, immunofluorescence was used to evaluate the effect of MCM on neuronal axon growth.
Luciferase assay
The wild-type (WT) or mutant-type (MUT) TUG1 3′-UTRs and TLR3 3′-UTRs were cloned into pmir-GLO plasmids. Compared with negative control (NC) mimics, miR-1192 mimics and TUG1 3′-UTRs or TLR3 3′-UTRs were co-transfected into 293T cells (National Collection of Authenticated Cell Culture, China, cat. no. GNHu17) with Lipofectamine 2000 (Invitrogen, USA). The Luciferase Reporter Assay System was used to detect luciferase activity.
Fluorescence in situ hybridization
The spinal cord tissues of mice from different days were collected for frozen sections, and an lncRNA FISH kit (GenePharma, Shanghai, China) was used for RNA fluorescence in situ hybridization. According to the instructions, the frozen sections were rehydrated, digested with protease K, denatured, hybridized with TUG1 nucleotide probe, stained with DAPI, and then observed under a fluorescence microscope.
Functional assessment
The Basso mouse scale (BMS) was used to evaluate the recovery of motor function in mice at 1, 3, 7, 14, and 28 days after injury. The footprint was used to evaluate the step length recovery of mice by gait analysis after 28 days of PBM treatment. Two researchers who did not participate in the experiment performed functional assessment.
Statistical analysis
All the experiments were repeated at least three times independently. The data statistics of this study were processed by GraphPad Prism software (8.3.0 version). Student’s t-test was used for comparison between two groups. One-way analysis of variance (ANOVA) with least significance difference post hoc analysis was used for comparison of three groups or more. The measured data are presented as mean ± standard deviation (SD). ImageJ software was used to perform optical density statistics, axon length measurement, and positive cell count. P < 0.05 was considered to be statistically significant.



留言 (0)