Signs of the disease and complications
Our cases confirm that SIOD is characterized by low renal and high extrarenal phenotypic variability. Short stature and proteinuric nephropathy are the typical hallmarks of SIOD and usually the first signs suggesting the disease. [5] All our patients were identified initially by nephrologists involved in the diagnosis of NS, which represents a very heterogeneous group of diseases. Determining the etiology of SRNS is crucial for the proper management of these patients. The proportion of genetic SRNS from all SRNS patients has been reported in 14–64% cases; our national study showed that 38% of children with SRNS have a confirmed genetic etiology [12]. Some of these children present with syndromic NS with typical specific extrarenal features that allow clinicians to establish an accurate diagnosis.
All children were born preterm with low birth weight, intrauterine growth retardation (IUGR), characteristic facial stigmatization (broad nasal bridge) and the majority of them had pigmented skin macules. Edema was the presenting sign in four children, and two presented with only nephrotic proteinuria. Renal biopsy was performed in three children; FSGS was found in two cases and IgM nephropathy was revealed in one child. Interestingly, based on our knowledge, our case is the first IgM nephropathy diagnosed in an SIOD patient reported to date. Generally, IgM nephropathy is an immune complex-mediated glomerulopathy characterized by diffuse mesangial immunoglobulin M deposits. Patients with IgM nephropathy are more often dependent on or resistant to corticosteroids. [13] However, histologic diagnosis is not decisive for the management of SIOD, so it may be avoided. Therefore, kidney histology results from many SIOD children are unavailable. Of note, four children from our cohort (80%) reached ESKD due to chronic progressive proteinuric nephropathy at the age ≤ 6 years.
The main complications and the major cause of mortality of SIOD patients are infections mainly associated with T-cell deficiency. Four patients from our cohort suffered from recurrent or protracted infections. We tried to identify the etiology of the infections and we treated them appropriately. Severe sepsis was the cause of death in case 1; moreover, we observed significant decompensation of the NS with generalized edema requiring albumin infusion and intensification of diuretic therapy during a COVID-19 infection in case 5.
Disproportionate short stature with short trunk is the result of spondyloepiphyseal dysplasia. An anthropometric study by Lücke et al. found that SIOD patients differed significantly from other individuals with CKD. The ratio of sitting height:leg length < 0.83 was specific for SIOD [14].
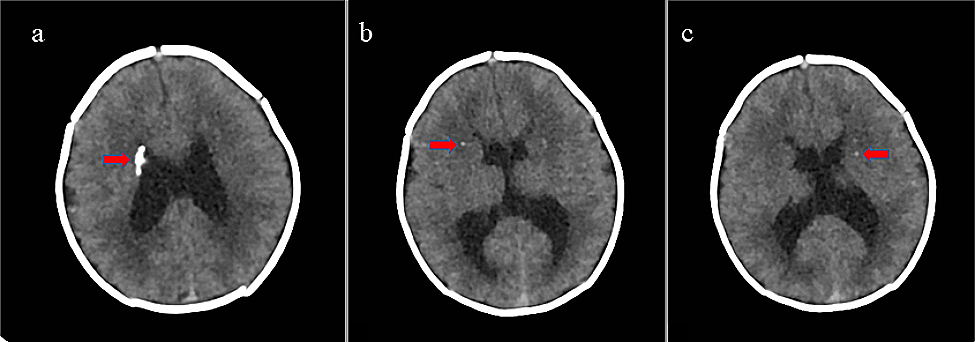
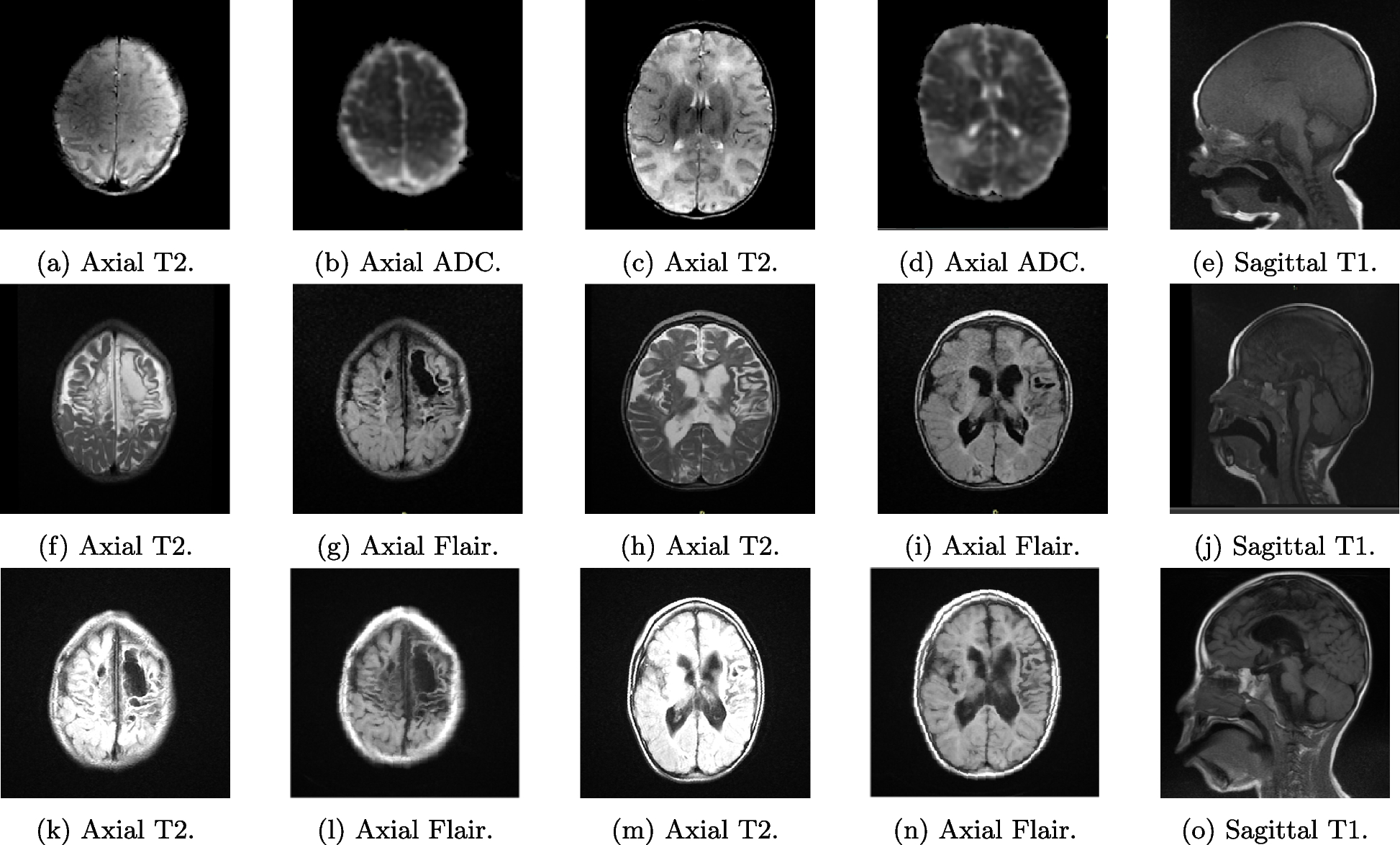
SMARCAL1 deficiency may lead to vascular brain disease; the resulting neurologic symptoms may be severe and can significantly reduce the quality of life. In some patients, the brain damage may be irreversible. Transient ischemic attacks (TIA) were first described by Ehrich et al. in three children who had perfusion defects revealed by positron emission tomography [15]. The expression of SMARCAL1 in brain tissue and the association of SMARCAL1 deficiency and abnormal brain development was reported later [16]. Kilic et al. hypothesized that SMARCAL1 may regulate the expression of genes necessary for neurological homeostasis. In addition, they postulated that disturbed expression of SMARCAL1 in the vasculature and smooth muscle may contribute to vascular dysfunction in SIOD [17]. Based on our current knowledge, reversible cerebral vasoconstriction syndrome, atherosclerosis and reduced elastogenesis contribute to the development of cerebral signs [18, 19]. The trigger of cerebral attacks may be blood pressure fluctuations, but these were not always present in our patients. Endothelial injury is also accentuated by CKD complications in SIOD individuals. In addition, management of hypertension in children treated with dialysis is challenging. We observed triplegia with expressive aphasia as a result of recurrent ischemic attacks in case 1. Cases 3 and 4 suffered from recurrent transient ischemic attacks, strokes and headaches. With time, the duration of attacks became longer, and symptoms were more pronounced. All our patients were treated by antiaggregation; we tried administering calcium channel blockers, minoxidil, beta blockers, paracetamol and non-steroidal anti-inflammatory drugs for neurologic symptoms with unsatisfactory effects. Unfortunately, no potent therapy for these complications has been established yet. Invasive intravascular treatment and thrombolysis are not recommended because of the age, body weight, abnormal tissue of the vessels and frequent recurrency of episodes.
Hematologic disease may occur during the course of SIOD disease. Leukopenia is a characteristic sign of the disease and was present in all our reported children. Evans syndrome resistant to multiple therapy including rituximab and bone marrow failure developed in case 1 [11]. In children with preserved diuresis, nephrotic proteinuria results in severe hypogammaglobulinemia, which aggravates the immunodeficient state in SIOD.
Genotype–phenotype correlation
Homozygous or compound heterozygous causal variants were identified in all our patients using next generation sequencing. SIOD is a disease with a variable phenotypic presentation. While spondyloepiphyseal dysplasia, dysmorphic features, renal disease and T-cell deficiency are typical features of SIOD, neurologic symptoms, autoimmune diseases and other organ symptoms are present in only a proportion of affected individuals. The pathogenic mechanisms leading to SIOD are not yet fully understood. Finding modifiers and other genes associated with SIOD is expected [20]. The phenotype varies from mild to severe disease; interestingly, the genotype to phenotype correlation is very weak, probably due to variable gene expression, oligogenic inheritance, epigenetics and environmental factors [3]. Lama et al. reported three siblings with different disease courses. While one boy died with severe nephropathy on hemodialysis at the age of 12 years, his brother survived into adulthood. He was started on hemodialysis when he was 22 years old and subsequently received a kidney graft. Their sister maintained normal kidney function throughout childhood [21].
Management
To date, no effective causal therapy for SIOD has been found. It is important to make the correct diagnosis early. Our reported children were diagnosed late and were exposed to unnecessary immunosuppressive therapy, which could facilitate the development of life-threatening infections. Nephropathy in SIOD is not amendable to immunosuppressive therapy. Steroids for manifestations of NS were used in all our patients before the correct diagnosis was established. Based on current knowledge, this therapy is of no value for genetic SRNS. Angiotensin-converting enzyme inhibitors may decrease proteinuria in these individuals; however, this has not been demonstrated by studies yet. Four patients from our cohort reached ESKD and PD was started. Lücke et al. published a case series of children with SIOD who successfully underwent kidney transplantation (Tx) with reduced immunosuppression due to T-cell immunodeficiency [6]. However, generally, there is a reluctance to perform kidney Tx because of the high risk of severe infections. On the other hand, long-term dialysis poses a threat of systemic side effects including progression of atherosclerosis, cardiovascular disease precipitating central nervous system disease and shortened life span. SIOD is and ultra-rare disease and clinical course of these individuals after kidney Tx could not have been studied well yet. [22] There is only limited evidence supporting kidney Tx in these children. Therefore, performing kidney Tx remains controversial in SIOD patients mainly due to the risk of life-threatening infections. Importantly, the majority of our patients have serious extrarenal symptoms, which may progress regardless of kidney Tx.
A special vaccination protocol for children with T-cell deficiency with the avoidance of live-attenuated vaccines was followed. Antibiotic prophylaxis (cotrimoxazole) was prescribed for SIOD patients to prevent opportunistic infection. [3] Although previous reports on bone marrow Tx have been rather disappointing, as four of five patients in the cohort did not survive [23], promising results have been published recently by Bertaina et al., who performed alpha/beta T-cell depleted haploidentical stem cell Tx followed by kidney Tx from a living donor. This revolutionary method lessens the risk of rejection and enables the minimization of immunosuppression with subsequent discontinuation of immunosuppressive medication. More experience with this promising therapeutic method is expected [24].
Limitations
The limitation of our study is the small sample size, but SIOD is an extremely rare disease and our study reports all the children presenting with this disease at our center over a period of 20 years.





留言 (0)